Separate cellNames based on biological variable during the creation of metacell aggregates #1199
Replies: 1 comment 3 replies
-
|
I cant comment on the exact code changes but the concept seems fine. Though I feel like you could accomplish this just by doing a project subset first. |
Beta Was this translation helpful? Give feedback.
-
|
By separating healthy v. disease cells in the metacell aggregate formation, I can compute the correlations across healthy and disease metacell aggregates. This analysis would be different from computing correlations using only healthy cells (subset 1) then computing correlations using only disease cells (subset 2). The former case is what I am trying to shoot for. I am currently assessing if this change is needed at all by counting the number of metacell aggregates that contain both healthy and diesease cells. If <5% of metacell aggregates are "healthy - disease mosaics" then it may not be a problem at all. I will keep you updated! Thanks |
Beta Was this translation helpful? Give feedback.
-
|
Update: Only ~7% of metacell aggregates contain both healthy and disease cells when running the default version of Therefore, this concern when running the default may not be as important as I thought. I suspect this would depend on the dataset and how LSI embeds healthy and disease cells in low dimensional space which could be affected by a number of factors (both biological and technical). |
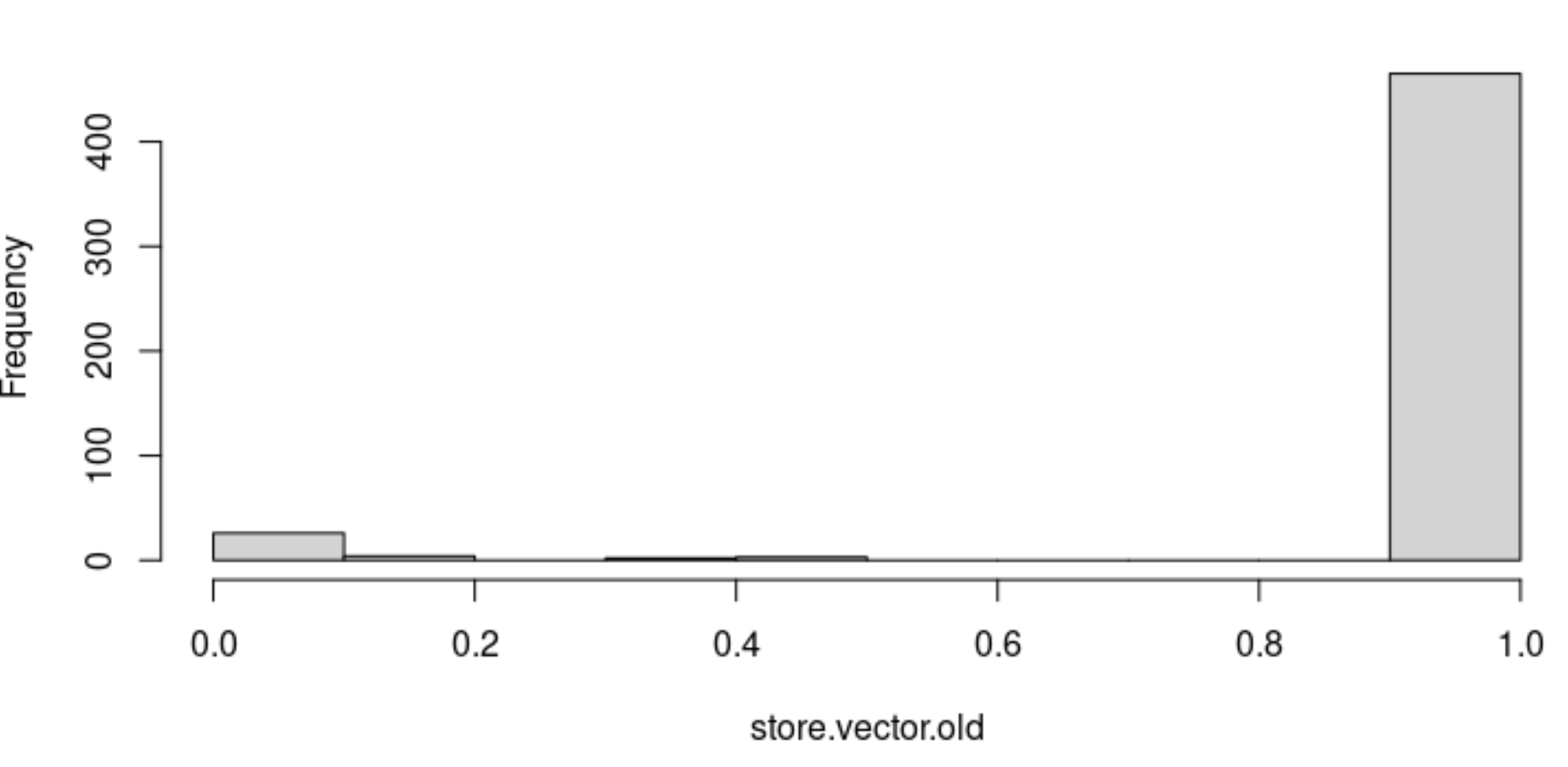
Beta Was this translation helpful? Give feedback.
-
|
interesting! Thanks for posting this! |
Beta Was this translation helpful? Give feedback.
Uh oh!
There was an error while loading. Please reload this page.
Uh oh!
There was an error while loading. Please reload this page.
-
Hi @rcorces and @jgranja24
I am working with a scATAC-seq dataset that includes both healthy and diseased tissue samples.
When performing peak-to-gene linkage and co-accessibility analyses, the single cell observations are aggregated into metacells via kNN to create more informative observations for computing correlations.
In this case, I think it is important for cells that make up a metacell aggregate to be all from the same biological condition/group. In other words, healthy cells should form healthy aggregates and disease cells should form disease aggregates. Creating metacells that include both healthy and disease cells could obscure the downstream results.
To ensure that the aggregate metacells contain only cells from one biological condition or group, I made some slight changes to the source code of
addCoAccessibilityandaddPeak2GeneLinks.I modified
addCoAccessibilitytoaddCoAccessibility.modby adding two additional parameters,group1andgroup2which are character vectors of cellNames (barcodes). In this case,group1is a vector of disease cellNames andgroup2is a vector of healthy cellNames. After thecellsToUseparameter is checked, I first subset the reducedDims to only the cells ofgroup1. Using thesegroup1cells, I proceed as normally by performing the subsampling for anidx, running the kNN, determining the overlaps in cells between metacell aggregates, and converting the knnObject toknnObj.group1. I then repeat these steps after subsetting the reducedDims to only the cells ofgroup2resulting in the knnObjectknnObj.group2. Finally, I concatenate these knnObj lists into one list that is used for construction of the metacell aggregate matrices.I implemented the same changes described above and modified
addPeak2GeneLinkstoaddPeak2GeneLinks.mod:Are these changes reasonable and accurate? Both modified functions successfully run to completation on my end. I would really appreciate your help and advice on this matter.
Beta Was this translation helpful? Give feedback.
All reactions