TSS assignment and p2g analysis #1313
-
|
Hi @rcorces Does assigning an alternative TSS for a gene influence the p2g correlation analysis? For a specific gene(NRG1) ArchR assigns the longest transcript as the main one for NRG1, and uses that transcripts' TSS. but when I look at NRG1, I see that another isoform is more expressed (the one circled) Even though the assigned TSS is different from what I visually see, the inferred expression of NRG1 is in the correct cluster (when I do the integration). I was wondering if the TSS assignment could potentially influence the peak2gene analysis in this case?
|
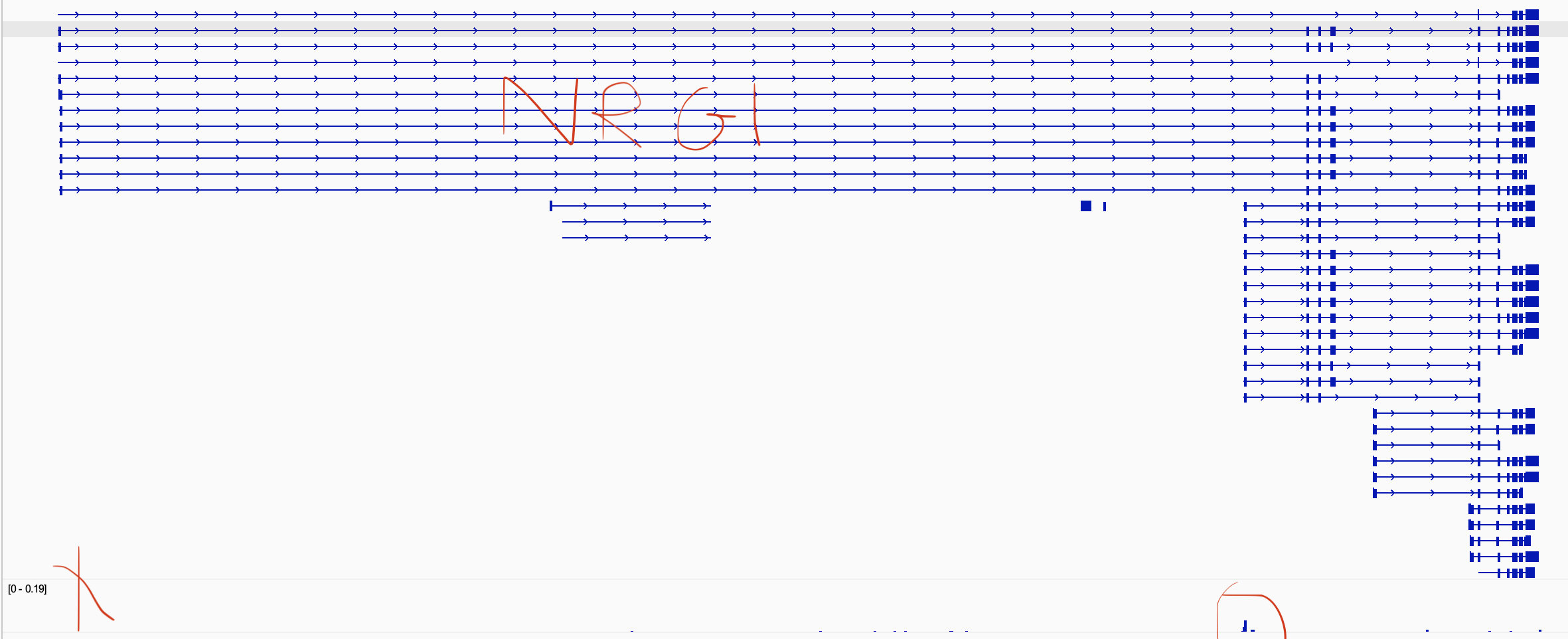
Beta Was this translation helpful? Give feedback.
Replies: 4 comments 2 replies
-
|
@rcorces I would very much appreciate it if I could get you advice into this matter, thank you so much again |
Beta Was this translation helpful? Give feedback.
-
|
I think since addReproduciblePeakSet uses promoter region (2000,100), if a TSS isn't assigned to the isoform of interest (the one with the highest accessibility at its TSS), this may influence the peak calling, and as a result that might influence the p2g analysis? |
Beta Was this translation helpful? Give feedback.
-
|
I'm sorry for not replying sooner. The genes of interest come from the geneAnnotation object. To keep analysis straightforward, only one start and stop is used for each gene. Not every TSS is used. So the positions being used for addPeakToGeneLinks are the positions of the genes shown by |
Beta Was this translation helpful? Give feedback.
-
|
Thanks @rcorces this might be a bug, I tried fixing NRG1 manually, I changed NRG1 start and end based on the isoform that I was interested in, and when I looked at the new grange it looks fine, I gave the new grange to addGeneScoreMatrix, and checked the genes again using getGenes, and I noticed that it hasn't been fixed(NRG1 start and end are not what I defined). Please let me know if this is a bug, I can post it in the issue section if it is, thanks again!
|


Beta Was this translation helpful? Give feedback.
-
|
I worked around the above issue by giving the new grange to the ArchR obj as follow: projHeme2@geneAnnotation$genes = getgenes0 Thanks again |
Beta Was this translation helpful? Give feedback.
-
|
That is expected.
Line 1061 in 968e442 where geneStart is the position used for p2g analysis.
In this case, it is taking the features from Line 248 in 968e442 So by changing the genes used in What I'm not sure about is if there will be any issues because your gene features dont match the genes in your |
Beta Was this translation helpful? Give feedback.
I'm sorry for not replying sooner. The genes of interest come from the geneAnnotation object. To keep analysis straightforward, only one start and stop is used for each gene. Not every TSS is used. So the positions being used for addPeakToGeneLinks are the positions of the genes shown by
getGenes(ArchRProj). If those dont fit your analysis, you would have to edit the gene annotation information and re-run the analysis (starting from GeneScoreMatrix creation).