Failing to identify subpopulation diversity #818
Replies: 3 comments 1 reply
-
|
Please note that the Issues section is meant for Bug reports, which this is not. I will migrate your issue to the Discussions section where it belongs. |
Beta Was this translation helpful? Give feedback.
-
|
We arent able to comment on user-specific analyses. If we did this, we would have a lot of similar requests. While we hope to improve the documentation in the future, the manual represents our advice as best as we can. Our hope is that one day other members of the community participate in the discussions posted here to help each other. |
Beta Was this translation helpful? Give feedback.
-
|
Okay, so focusing on non user specific analysis, how do you propose interrogating the features (bins/peaks) selected for clustering in order to QC and debug issues? Surely it's a basic mechanism that every user is required to do if they want to understand their data. |
Beta Was this translation helpful? Give feedback.
-
|
You could try to extract this information from the SVD matrix (stored in the |
Beta Was this translation helpful? Give feedback.
Uh oh!
There was an error while loading. Please reload this page.
-
I've been analyzing various mouse immune cell 10x scATACseq datasets and found rich diversity of myeloid cells using ArchR clustering, but so far the same strategy fails when applied to human immune cells although the sequencing/data quality was good -- I was wondering if you can point me to strategies/ArchR functionality to identify the source of the problem.
The specific issue is that myeloid subsets (dendritic cells, macrophages and monocytes) don't appear to segregate into distinct clusters as I saw before in other contexts.
This is the relevant code to my initial clustering strategy:
which I then refined by performing peak calling and using the peak matrix for feature selection, which didn't help by much.
Based on the top differentially accessible genes by gene score I do find some of the relevant markers popping up such as C1QC for macrophages, but it's non specific and seems to be mixed with dendritic cell markers like CD1C.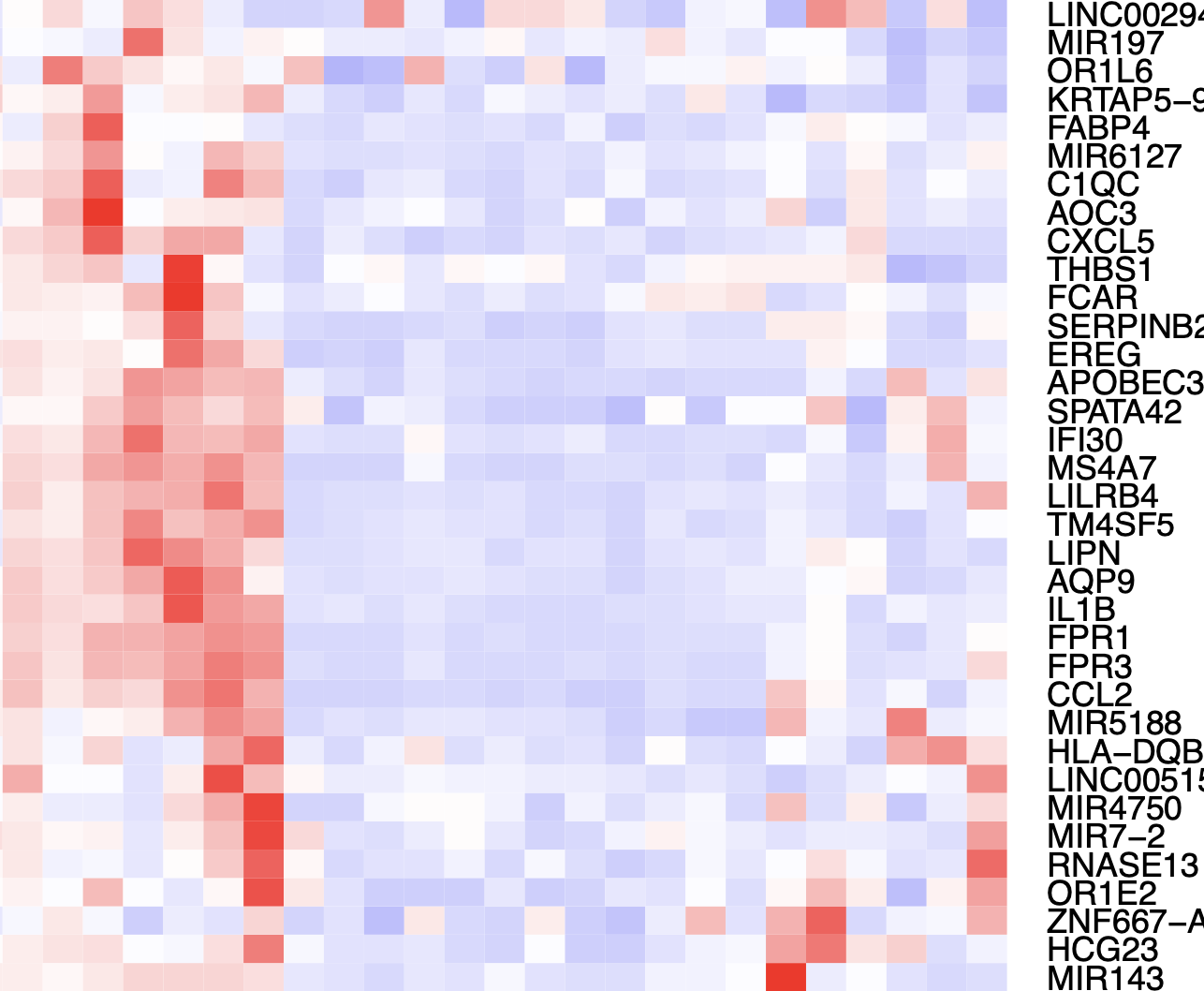
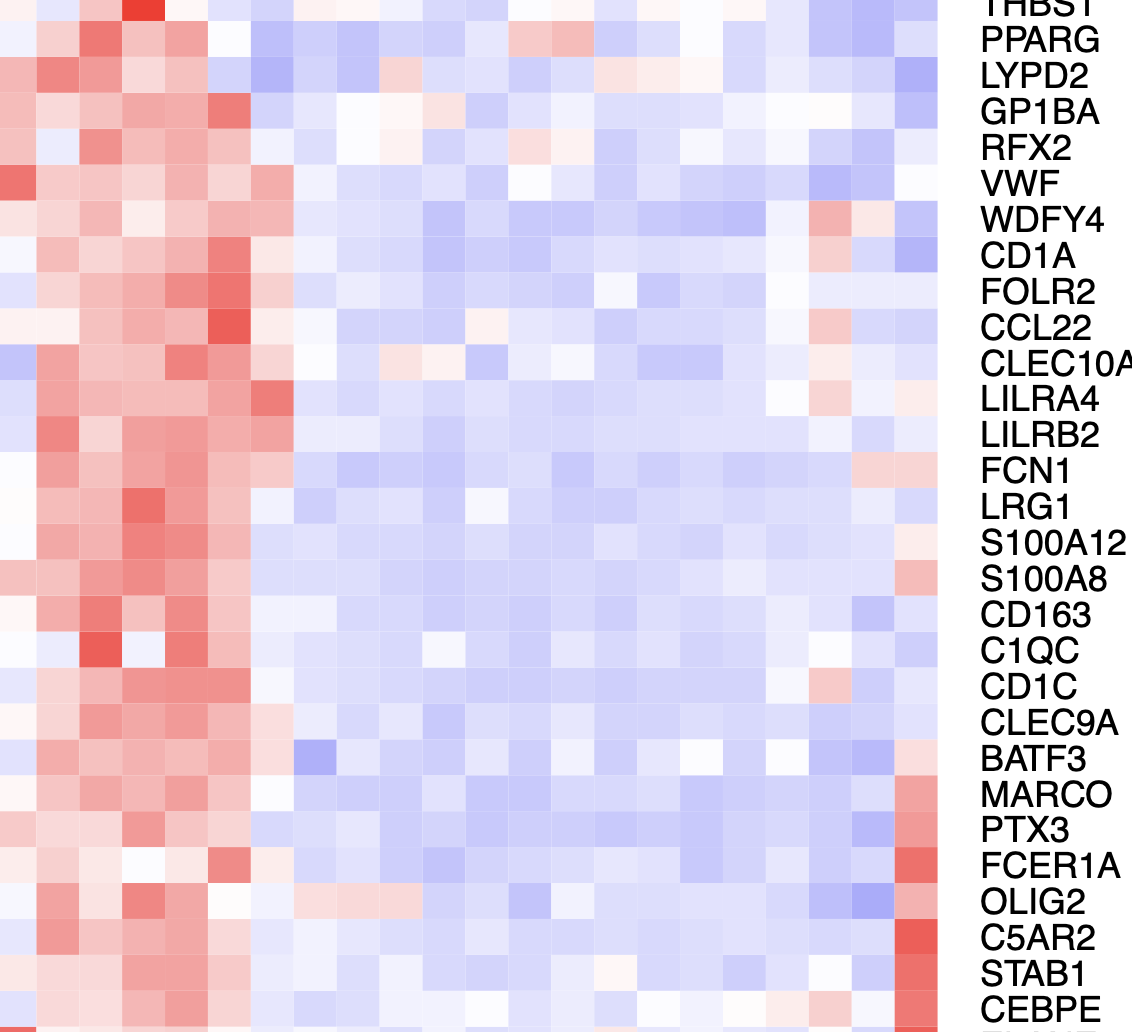
Some shared features might be driving them together like IL1B which is a known inflammatory feature, but also a lot of MIR/LINC genes pop up everywhere and I'm not sure if they confound the clustering and results?
Top differential genes:
Selected marker genes:
How do I interrogate this problem? Whether the features relevant to distinguishing these cell types? How do I extract the features used for clustering and annotate them by closest gene? Any other strategies to identify the source of this issue?
Thank you!
Cc'ing my colleague @s7hegde
Beta Was this translation helpful? Give feedback.
All reactions