Custom peakAnnotation for ENCODE data #916
Replies: 2 comments 1 reply
-
I think you have misunderstood the parameter documentation
In the manual, we DEFINE a
Overlap is any single base of overlap.
I dont understand this question. But in general, we dont respond to questions about user-specific how to get a specific plot or heatmap because we do not have bandwidth. The answer will likely be that you need to take the output from ArchR and plot the heatmap using code that you develop. |
Beta Was this translation helpful? Give feedback.
-
|
Thanks for the response. Just to clarify: So "ChIP" is just a place-holder name that should match the name in the addPeakAnnotations custom regions? Or is it actually pointing out to a specific type of custom code that assumes data as ChiP? The DNAse files are also in bed format so if that's all what matters, the ni could just choose the name as DNAse and should run it. Thanks for clarification on this matter. |
Beta Was this translation helpful? Give feedback.
-
|
This information is all contained within the manual and function documentation. Please explore these extensively before posting. From the manual: From the function documentation: |
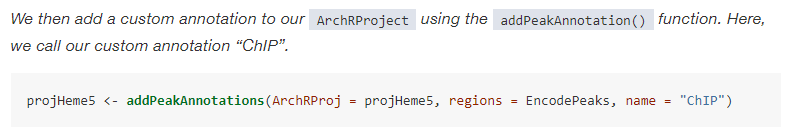
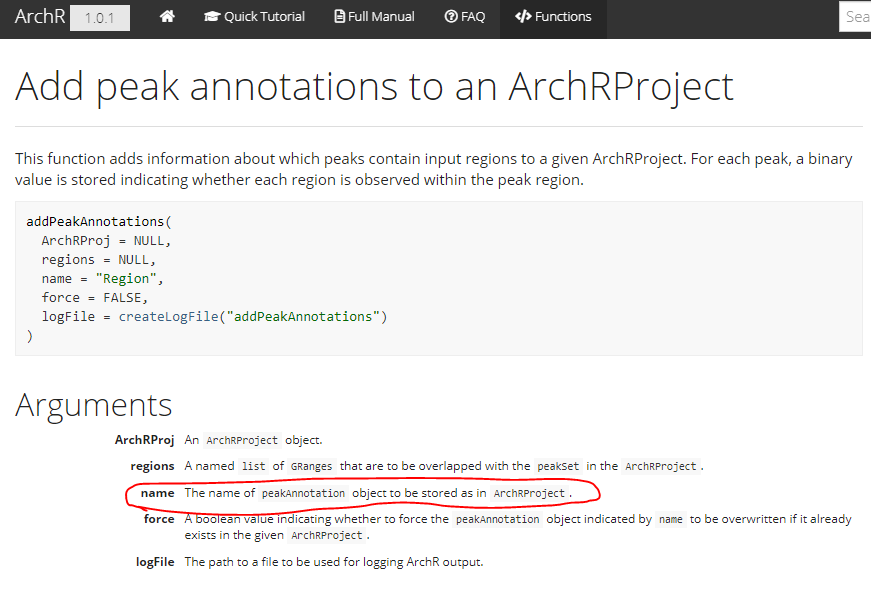
Beta Was this translation helpful? Give feedback.
Uh oh!
There was an error while loading. Please reload this page.
-
Thanks so much for providing this pipeline where Encode data can be interrogated in a user-friendly manner. Few questions while testing the the Custom Enrichment https://www.archrproject.com/bookdown/custom-enrichment.html
The peakAnnoEnrichment function option for peakAnnotation = "ChiP" works for ChiP data as shown in the exam n the manual. However, what if I am trying to use bed files from DNAse-seq data such as https://www.encodeproject.org/experiments/ENCSR328UMC/ : should I still use peakAnnotation = "ChiP"? The ?peaAnnotation help doesn't provide option selection for DNAse.
I couldn't find in the manual information on the parameters of the accessible peak location that would consider it an 'overlap'. What's the allowed deviation from the ENCODE database overlapping with the scATAC data that would still count it as an overlapping peak?
Is there a way to convert the overlapping peaks between our clusters/celltypes of interest with the ENCODE and convert that immediately to genes within ArchR to export as a heat map of overlapping genes between each of our clusters and the Encode custom file?
Thanks much for your input.
Beta Was this translation helpful? Give feedback.
All reactions