You signed in with another tab or window. Reload to refresh your session.You signed out in another tab or window. Reload to refresh your session.You switched accounts on another tab or window. Reload to refresh your session.Dismiss alert
Copy file name to clipboardExpand all lines: README.md
+39-20Lines changed: 39 additions & 20 deletions
Display the source diff
Display the rich diff
Original file line number
Diff line number
Diff line change
@@ -20,7 +20,7 @@ If you cannot wait to start codding, try to run some scripts from `examples` fol
20
20
## Basic Components
21
21
There are three basic components required for any simulation of systems of N-bodies: `bodies`, `system` and `simulation`.
22
22
23
-
**Bodies** or **Particles** are the objects which will interact with each other and for wich the equations of Newton's 2nd law are solved during the simulation process. Three parametes of a body are necessary, they are initial location, initial velocity and mass. `MassBody` structure represents such particles:
23
+
**Bodies** or **Particles** are the objects which will interact with each other and for which the equations of Newton's 2nd law are solved during the simulation process. Three parameters of a body are necessary, they are initial location, initial velocity and mass. `MassBody` structure represents such particles:
24
24
25
25
```julia
26
26
using StaticArrays
@@ -29,7 +29,7 @@ v = SVector(.1,.2,.5)
29
29
mass =1.25
30
30
body =MassBody(r,v,mass)
31
31
```
32
-
For the sake of simulation speed it is advised to use static arrays from the corresponding package.
32
+
For the sake of simulation speed it is advised to use [static arrays](https://github.com/JuliaArrays/StaticArrays.jl).
33
33
34
34
A **System** covers bodies and necessary parameters for correct simulation of interaction between particles. For example, to create an entity for a system of gravitationally interacting particles, one needs to use `GravitationalSystem` constructor:
There are different types of bodies but they are just containers of particle parameters. The interaction and acceleration of particles are defined by the potentials or force fields.
49
49
50
50
## Generating bodies
51
-
The package exports quite a useful function for placing simular particles in the nodes of a cubic cell with their velocities distributed in accordance with the Maxwell–Boltzmann law:
51
+
The package exports quite a useful function for placing similar particles in the nodes of a cubic cell with their velocities distributed in accordance with the Maxwell–Boltzmann law:
The Lennard-Jones potential is used in molecular dynamics simulations for approximating interactions between neutral atoms or molecules. The SPC/Fw water model is used in water simulations. The meaning of arguments for `SPCFwParameters` constructor will be clarified further in this documentation.
80
+
The Lennard-Jones potential is used in molecular dynamics simulations for approximating interactions between neutral atoms or molecules. The [SPC/Fw water model](http://www.sklogwiki.org/SklogWiki/index.php/SPC/Fw_model_of_water) is used in water simulations. The meaning of arguments for `SPCFwParameters` constructor will be clarified further in this documentation.
81
81
82
82
`PotentialNBodySystem` structure represents systems with a custom set of potentials. In other words, the user determines the ways in which the particles are allowed to interact. One can pass the bodies and parameters of interaction potentials into that system. In case the potential parameters are not set, during the simulation particles will move at constant velocities without acceleration.
83
83
@@ -90,7 +90,7 @@ There exists an [example](http://docs.juliadiffeq.org/latest/models/physical.htm
90
90
91
91
Here is shown how to create custom acceleration functions using tools of NBodySimulator.
92
92
93
-
First of all it is necessary to create a structure for parameters for the custom potential.
93
+
First of all, it is necessary to create a structure for parameters for the custom potential.
Interaction between charged particles obeys Coulomb's law. The movement of such bodies can be simulated using `ChargedParticle` and `ChargedParticles` structures.
161
161
162
-
The following example shows how to model two oppositely charged particles. If one body is more massive that another, it will be possible to observe rotation of the light body around the heavy one without ajusting their positions in space. The constructor for `ChargedParticles` system requires bodies and Coulomb's constant `k` to be passed as arguments.
162
+
The following example shows how to model two oppositely charged particles. If one body is more massive that another, it will be possible to observe rotation of the light body around the heavy one without adjusting their positions in space. The constructor for `ChargedParticles` system requires bodies and Coulomb's constant `k` to be passed as arguments.
An N-body system consisting of `MagneticParticle`s can be used for simulation of interacting magnetic dipoles, though such dipoles cannot rotate in space. Such a model can represent single domain particles interacting under the influence of a strong external magnetic field.
181
181
182
-
In order to create a magnetic particle one specifies its location in space, velocity and the vector of its magnetic moment. The following code shows how we can construct an iron particle:
182
+
In order to create a magnetic particle, one specifies its location in space, velocity and the vector of its magnetic moment. The following code shows how we can construct an iron particle:
183
183
184
184
```julia
185
185
iron_dencity =7800# kg/m^3
@@ -219,7 +219,7 @@ NBodySimulator allows one to conduct molecular dynamic simulations for the Lenna
219
219
The comprehensive examples of liquid argon and water simulations can be found in `examples` folder.
220
220
Here only the basic principles of the molecular dynamics simulations using NBodySimulator are presented using liquid argon as a classical MD system for beginners.
221
221
222
-
First of all one needs to define paramters of the simultaion:
222
+
First of all, one needs to define parameters of the simulation:
The default boundary conditions are `InfiniteBox` withouth any limits, default thermostat is `NullThermostat` which does no thermostating and default Boltzmann constant `kb` equals its value in SI, i.e. 1.38e-23 J/K.
267
+
The default boundary conditions are `InfiniteBox` without any limits, default thermostat is `NullThermostat` which does no thermostating and default Boltzmann constant `kb` equals its value in SI, i.e. 1.38e-23 J/K.
268
+
269
+
## Water Simulations
270
+
In NBodySImulator the [SPC/Fw water model](http://www.sklogwiki.org/SklogWiki/index.php/SPC/Fw_model_of_water) is implemented. For using this model, one has to specify parameters of the Lennard-Jones potential between the oxygen atoms of water molecules, parameters of the electrostatic potential for the corresponding interactions between atoms of different molecules and parameters for harmonic potentials representing bonds between atoms and the valence angle made from bonds between hydrogen atoms and the oxygen one.
water =WaterSPCFw(bodies, mH, mO, qH, qO, jl_parameters, e_parameters, spc_paramters);
278
+
```
279
+
280
+
For each water molecule here, `rOH` is the equilibrium distance between a hydrogen atom and the oxygen atom, `∠HOH` denotes the equilibrium angle made of those two bonds, `k_bond` and `k_angle` are the elastic coefficients for the corresponding harmonic potentials.
281
+
282
+
Further, one pass the water system into `NBodySimulation` constructor as a usual system of N-bodies.
Usually during simulation of a system is required to be at a particular temperature. NBodySimulator contains several thermostas for that purpose. Here the thermostating of liquid argon is presented, for thermostating of water one can refer to [this post](https://mikhail-vaganov.github.io/gsoc-2018-blog/2018/08/06/thermostating.html)
289
+
Usually during simulation of a system is required to be at a particular temperature. NBodySimulator contains several thermostats for that purpose. Here the thermostating of liquid argon is presented, for thermostating of water one can refer to [this post](https://mikhail-vaganov.github.io/gsoc-2018-blog/2018/08/06/thermostating.html)
Once the simulation is completed, one can analyze the result and obtain some useful characteristics of the system.
304
323
305
-
Function `run_simulation` returns a structure containig the initial parameters of simulation and the solution of differential equation (DE) required for description of the corresponding system of particles. There are different functions which help to intepret solution of DEs into physical quantities.
324
+
Function `run_simulation` returns a structure containing the initial parameters of simulation and the solution of differential equation (DE) required for description of the corresponding system of particles. There are different functions which help to interpret solution of DEs into physical quantities.
306
325
307
-
One of the main charachterestics of a system during molecular dynamics simulations is its thermodynamic temperature. The value of the temperature at a particular time `t` can be obtained via calling this function:
326
+
One of the main characteristics of a system during molecular dynamics simulations is its thermodynamic temperature. The value of the temperature at a particular time `t` can be obtained via calling this function:
308
327
309
328
```julia
310
329
T =temperature(result, t)
@@ -314,7 +333,7 @@ T = temperature(result, t)
314
333
The RDF is another popular and essential characteristic of molecules or similar systems of particles. It shows the reciprocal location of particles averaged by the time of simulation.
315
334
316
335
```julia
317
-
(rs, grf) =@timerdf(result)
336
+
(rs, grf) =rdf(result)
318
337
```
319
338
320
339
The dependence of `grf` on `rs` shows radial distribution of particles at different distances from an average particle in a system.
@@ -325,14 +344,14 @@ Here the radial distribution function for the classic system of liquid argon is
325
344
### Mean Squared Displacement (MSD)
326
345
The MSD characteristic can be used to estimate the shift of particles from their initial positions.
327
346
```julia
328
-
(ts, dr2) =@timemsd(result)
347
+
(ts, dr2) =msd(result)
329
348
```
330
-
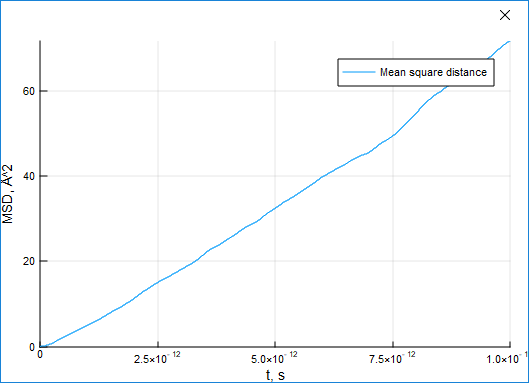
For a standrad liquid argon system the displacement grows with time:
349
+
For a standard liquid argon system the displacement grows with time:
331
350
332
351
333
352
### Energy Functions
334
353
335
-
Energy is a higlhy important physical characteristic of a system. The module provides four functions to obtain it, though the `total_energy` function just sums potential and kinetic energy:
354
+
Energy is a highly important physical characteristic of a system. The module provides four functions to obtain it, though the `total_energy` function just sums potential and kinetic energy:
Using tools of NBodySimulator one can export results of simulation into a [Protein Database File](https://en.wikipedia.org/wiki/Protein_Data_Bank_(file_format)). [VMD](http://www.ks.uiuc.edu/Research/vmd/) is a well-known tool for visualizing molecular dynamics, which can read data from PDB files.
346
365
347
366
```julia
@@ -358,4 +377,4 @@ plot(result)
358
377
animate(result, "path_to_file.gif")
359
378
```
360
379
361
-
Makie.jl also has a recipe for plotting results of N-body simulations. The [example](http://makie.juliaplots.org/stable/examples-meshscatter.html#Type-recipe-for-molecule-simulation-1) is presenten in the documentation.
380
+
Makie.jl also has a recipe for plotting results of N-body simulations. The [example](http://makie.juliaplots.org/stable/examples-meshscatter.html#Type-recipe-for-molecule-simulation-1) is presented in the documentation.
0 commit comments