diff --git a/source/_posts/unimol_deepg.md b/source/_posts/unimol_deepg.md
new file mode 100644
index 00000000..c3e403a3
--- /dev/null
+++ b/source/_posts/unimol_deepg.md
@@ -0,0 +1,101 @@
+---
+title: "What Can Uni-Mol Do too? | Unveiling DeepGlycanSite: Precise Prediction of Carbohydrate Binding Sites"
+date: 2024-07-22
+categories:
+- Uni-Mol
+mathjax: true
+---
+
+On June 17, 2024, researchers Xi Cheng and Liuqing Wen from the Shanghai Institute of Materia Medica, Chinese Academy of Sciences, in collaboration with Dingyan Wang from Lingang Laboratory, published a study titled *"Highly accurate carbohydrate-binding site prediction with DeepGlycanSite"* in *Nature Communications* [1]. This research introduces DeepGlycanSite, a deep learning-based algorithm for predicting carbohydrate-binding sites on protein structures with high precision. By leveraging Uni-Mol, DeepGlycanSite achieves exceptional accuracy in identifying carbohydrate-binding sites, providing a powerful tool for studying carbohydrate-protein interactions.
+
+## 1. Research Background
+Carbohydrates are widely present on the surface of all living cells, interacting with various protein families, including lectins, antibodies, enzymes, and transport proteins. These interactions regulate diverse biological processes, such as immune responses, cell differentiation, and neural development. Understanding carbohydrate-protein interactions is therefore fundamental to developing carbohydrate-based therapeutics.
+
+However, due to the structural diversity of carbohydrates, obtaining experimental data on carbohydrate-protein interactions remains challenging. Structural determination techniques commonly used in glycobiology, such as nuclear magnetic resonance (NMR) and X-ray crystallography, require pure, stable molecules of detectable sizes.
+
+Small carbohydrates (e.g., glucose with a molecular weight under 200 Da) are difficult to detect in structural studies due to their low atom count. On the other hand, complex long-chain carbohydrates (e.g., oligosaccharides with molecular weights exceeding 1000 Da) often involve multiple conformational states, leading to heterogeneity. In both cases, carbohydrate-binding residues of proteins cannot be clearly defined from a structural perspective.
+
+Thus, developing a reliable tool for predicting carbohydrate-binding sites is critical to advancing our understanding of carbohydrate-protein interactions.
+
+## 2. **Cutting-Edge Deep Learning Technology—DeepGlycanSite**
+
+DeepGlycanSite is an equivariant graph neural network (EGNN) model based on deep learning, combining geometric features of proteins with evolutionary information to outperform state-of-the-art methods. This model not only predicts binding sites for monosaccharides and disaccharides but also accurately identifies binding sites for oligosaccharides and nucleotides.
+
+The success of this study lies in the precise understanding of carbohydrate chemical structures, a capability significantly enhanced by Uni-Mol, which plays a critical role in the model's performance.
+
+ +
+
+## 3. **How Does Uni-Mol Assist DeepGlycanSite?**
+
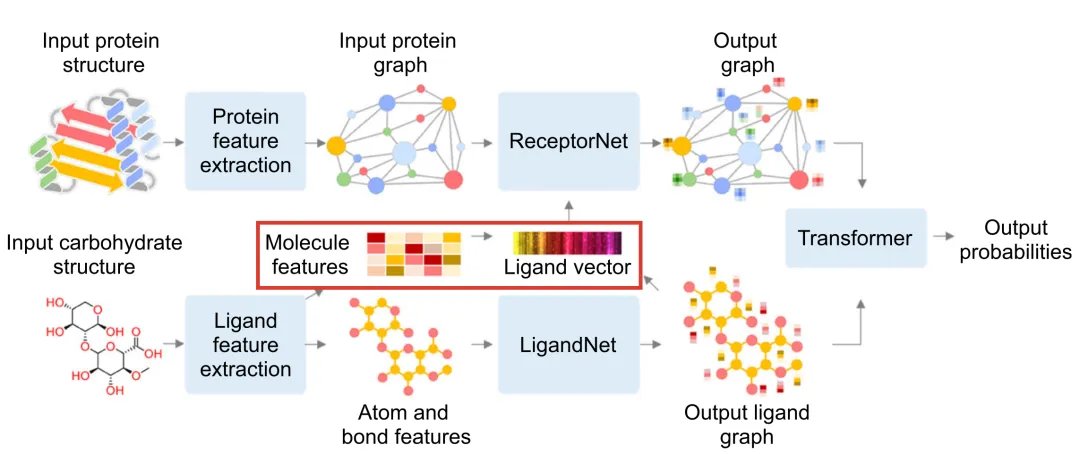
+The performance of deep learning models heavily depends on the quality of feature extraction. In DeepGlycanSite, Uni-Mol is utilized to generate detailed chemical features of carbohydrates, enabling more accurate prediction of binding sites. The implementation is as follows:
+
+---
+
+### **3.1 Carbohydrate Processing**
+- **SMILES Representation**:
+ Rdkit is used to process the query carbohydrate and extract its SMILES representation.
+- **Feature Generation**:
+ Uni-Mol, integrated with Rdkit, converts the SMILES representation into molecular features.
+
+---
+
+### **3.2 Feature Extraction**
+
+#### **Node Features**:
+Include detailed atomic properties:
+- Atom symbol
+- Degree
+- Hybridization type
+- Formal charge
+- Number of radical electrons
+- Aromaticity
+- Total hydrogen count
+- Chirality
+
+#### **Edge Features**:
+Capture bond-level information:
+- Bond type
+- Conjugation
+- Ring membership
+- Stereochemical configuration
+
+#### **Global Molecular Features**:
+Generate a 512-dimensional molecular feature vector encapsulating the overall chemical information of the carbohydrate.
+
+---
+
+### **3.3 Feature Integration**
+
+In the **DeepGlycanSite+Ligand** module:
+- The ligand vector generated by Uni-Mol is fused with the protein graph’s node features.
+- This integration is processed through an attention layer for feature alignment and updating.
+- The combined features are then used to predict the binding probability of carbohydrates.
+
+
+
+
+## 3. **How Does Uni-Mol Assist DeepGlycanSite?**
+
+The performance of deep learning models heavily depends on the quality of feature extraction. In DeepGlycanSite, Uni-Mol is utilized to generate detailed chemical features of carbohydrates, enabling more accurate prediction of binding sites. The implementation is as follows:
+
+---
+
+### **3.1 Carbohydrate Processing**
+- **SMILES Representation**:
+ Rdkit is used to process the query carbohydrate and extract its SMILES representation.
+- **Feature Generation**:
+ Uni-Mol, integrated with Rdkit, converts the SMILES representation into molecular features.
+
+---
+
+### **3.2 Feature Extraction**
+
+#### **Node Features**:
+Include detailed atomic properties:
+- Atom symbol
+- Degree
+- Hybridization type
+- Formal charge
+- Number of radical electrons
+- Aromaticity
+- Total hydrogen count
+- Chirality
+
+#### **Edge Features**:
+Capture bond-level information:
+- Bond type
+- Conjugation
+- Ring membership
+- Stereochemical configuration
+
+#### **Global Molecular Features**:
+Generate a 512-dimensional molecular feature vector encapsulating the overall chemical information of the carbohydrate.
+
+---
+
+### **3.3 Feature Integration**
+
+In the **DeepGlycanSite+Ligand** module:
+- The ligand vector generated by Uni-Mol is fused with the protein graph’s node features.
+- This integration is processed through an attention layer for feature alignment and updating.
+- The combined features are then used to predict the binding probability of carbohydrates.
+
+ +
+## 4. **Experimental Validation and Results**
+
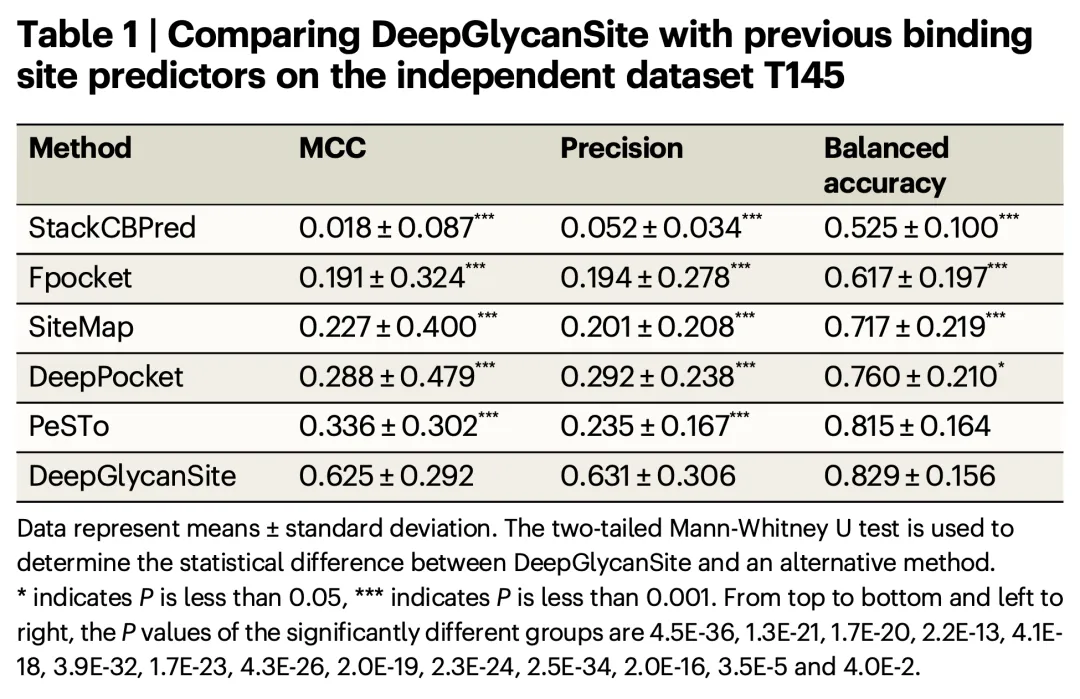
+The study constructed a large dataset containing approximately 8,100 proteins and 1,700 carbohydrates and evaluated the performance of the DeepGlycanSite model on multiple independent test sets. The results demonstrated that DeepGlycanSite outperforms existing methods in detecting carbohydrate-binding sites.
+
+- **Key Metrics**:
+ - **Matthew’s Correlation Coefficient (MCC)**: 0.625 (average on independent test sets)
+ - **Precision**: 0.631
+ - **Balanced Accuracy**: 0.829
+
+These metrics significantly exceed those of other comparison methods, highlighting the superior performance of DeepGlycanSite.
+
+
+
+## 4. **Experimental Validation and Results**
+
+The study constructed a large dataset containing approximately 8,100 proteins and 1,700 carbohydrates and evaluated the performance of the DeepGlycanSite model on multiple independent test sets. The results demonstrated that DeepGlycanSite outperforms existing methods in detecting carbohydrate-binding sites.
+
+- **Key Metrics**:
+ - **Matthew’s Correlation Coefficient (MCC)**: 0.625 (average on independent test sets)
+ - **Precision**: 0.631
+ - **Balanced Accuracy**: 0.829
+
+These metrics significantly exceed those of other comparison methods, highlighting the superior performance of DeepGlycanSite.
+
+ +
+
+### **Conclusion**
+
+DeepGlycanSite is a highly efficient prediction tool that leverages Uni-Mol’s robust molecular representation capabilities to enhance the accuracy of carbohydrate-binding site predictions on proteins. By integrating sequence and structural information, DeepGlycanSite not only surpasses traditional methods in detecting monosaccharide or disaccharide binding sites but also excels in identifying multiple binding sites. This provides critical insights into carbohydrate-protein interactions.
+
+Uni-Mol's ability to precisely capture chemical features and significantly improve predictive performance has established DeepGlycanSite as a powerful tool for addressing complex biological tasks. Its low dependence on protein structural accuracy enables analysis using predicted structures, supporting research into carbohydrate biological functions and drug development.
+
+The study encourages researchers to explore Uni-Mol for various downstream applications in different domains. The team welcomes collaboration and discussion to unlock further possibilities!
+
+Reference:
+[1] He, X., Zhao, L., Tian, Y. et al. Highly accurate carbohydrate-binding site prediction with DeepGlycanSite. Nat Commun 15, 5163 (2024). https://doi.org/10.1038/s41467-024-49516-2
+[2] Zhou G, Gao Z, Ding Q, Zheng H, Xu H, Wei Z, et al. Uni-Mol: A Universal 3D Molecular Representation Learning Framework. ChemRxiv. 2023; doi:10.26434/chemrxiv-2022-jjm0j-v4
\ No newline at end of file
diff --git a/source/_posts/unimol_kpi.md b/source/_posts/unimol_kpi.md
new file mode 100644
index 00000000..8770cc6a
--- /dev/null
+++ b/source/_posts/unimol_kpi.md
@@ -0,0 +1,84 @@
+---
+title: "What Can Uni-Mol Do too? | Predicting the molecular properties of battery electrolytes with the integrated KPI framework"
+date: 2024-12-02
+categories:
+- Uni-Mol
+mathjax: true
+---
+
+With the growing market demand for efficient and safe rechargeable batteries that can operate under extreme temperature conditions, the rapid and accurate evaluation of key properties of electrolyte molecules has become particularly important. A recent paper titled "A Knowledge–Data Dual-Driven Framework for Predicting the Molecular Properties of Rechargeable Battery Electrolytes," published in Angewandte Chemie International Edition, details an innovative approach known as the "Knowledge–Data Dual-Driven Framework" (KPI) specifically designed to predict the molecular properties of battery electrolytes, including melting point (MP), boiling point (BP), and flash point (FP). The research team skillfully combined deep learning techniques with domain-specific chemical knowledge, supported by large-scale datasets, significantly enhancing the accuracy and efficiency of predictions. In this framework, the Uni-Mol model plays a central role, demonstrating great potential in predicting the properties of electrolyte molecules and providing strong support for the development of next-generation high-performance batteries.
+
+Xiang Chen, an associate research fellow in the Department of Chemical Engineering at Tsinghua University, is the corresponding author of the paper. Yuchen Gao, a 2022 direct-entry PhD student in the Department of Chemical Engineering, is the first author. Co-authors include Yuhang Yuan, an undergraduate from the Tsinghua Academy of Wisdom; Suozhi Huang, an undergraduate from the Institute for Interdisciplinary Information Sciences; Nan Yao, a 2020 direct-entry PhD student; Legeng Yu, a 2021 direct-entry PhD student; Yaopeng Chen, a 2023 direct-entry PhD student; and Qiang Zhang, a professor in the Department of Chemical Engineering. The research was supported by funding from the National Natural Science Foundation of China, the National Key R&D Program of China, and the Beijing Natural Science Foundation.
+
+
+
+## 1. Research Background
+
+Secondary batteries have been widely used in electric vehicles and portable electronic devices. However, with the diversification of application scenarios—such as extreme cold, desert regions, and specialized settings like aerospace, underground exploration, and medical device sterilization—the demand for batteries to maintain stable performance under extreme temperatures has become increasingly urgent.
+
+In high-temperature environments, side reactions between electrolytes and active materials intensify, not only consuming electrolytes and active substances but also potentially triggering thermal runaway, posing serious safety risks. Conversely, in low-temperature conditions, the kinetics of electrochemical reactions are significantly reduced, which can lead to the formation of lithium dendrites, further increasing safety hazards. Therefore, developing batteries capable of safe operation across a wide temperature range has become a critical technical challenge.
+
+Moreover, battery performance heavily depends on the physicochemical properties of electrolytes, such as melting point, boiling point, and flash point. Traditional experimental methods, however, are inefficient due to limited data and reliance on trial-and-error processes, making them inadequate for the rapid development of new electrolyte molecules. Against this backdrop, efficient molecular property prediction models and high-throughput screening tools are particularly important to support the advancement of battery research toward higher performance and enhanced safety.
+
+## 2. Methods
+In this paper, the research team developed an innovative approach called the Knowledge–Data Dual-Driven Framework (KPI) for predicting the molecular properties of secondary battery electrolytes. The KPI framework consists of three main modules: data organization and statistical analysis, interpretability and knowledge discovery, and knowledge-based molecular property prediction. First, the KPI framework collects molecular structure and property data from public databases and literature and automatically organizes it into structured datasets. Next, it employs interpretable machine learning methods to explore structure–property relationships from a microscopic perspective. Finally, the discovered knowledge is embedded into property prediction models.
+
+Uni-Mol, serving as a foundational model, is utilized for molecular property prediction within the framework. It integrates a knowledge embedding mechanism to enhance prediction performance through pretraining and fine-tuning.
+
+1. **Pretraining**:
+ Uni-Mol undergoes pretraining on a large dataset of chemical molecules (in SMILES notation) to learn the underlying relationships between molecular structures and properties. This process provides a robust molecular representation foundation for property prediction.
+
+2. **Knowledge Embedding and Fine-Tuning**:
+ For specific tasks (e.g., predicting melting point, boiling point, and flash point), Uni-Mol integrates critical information extracted from data and chemical knowledge through a knowledge embedding mechanism. This mechanism uses a knowledge purity controller and a knowledge flow controller to adjust the proportion and dimensionality of embedded knowledge vectors, significantly improving the model's prediction accuracy.
+
+3. **Efficient Screening and Molecular Design (KPI-Assisted Electrolyte Molecular Design)**:
+ The fine-tuned Uni-Mol achieves high accuracy in predicting molecular properties, enabling high-throughput screening to quickly identify molecules with excellent performance. This supports the development of wide-temperature-range and high-safety electrolytes.
+
+## 3. Results and Discussion
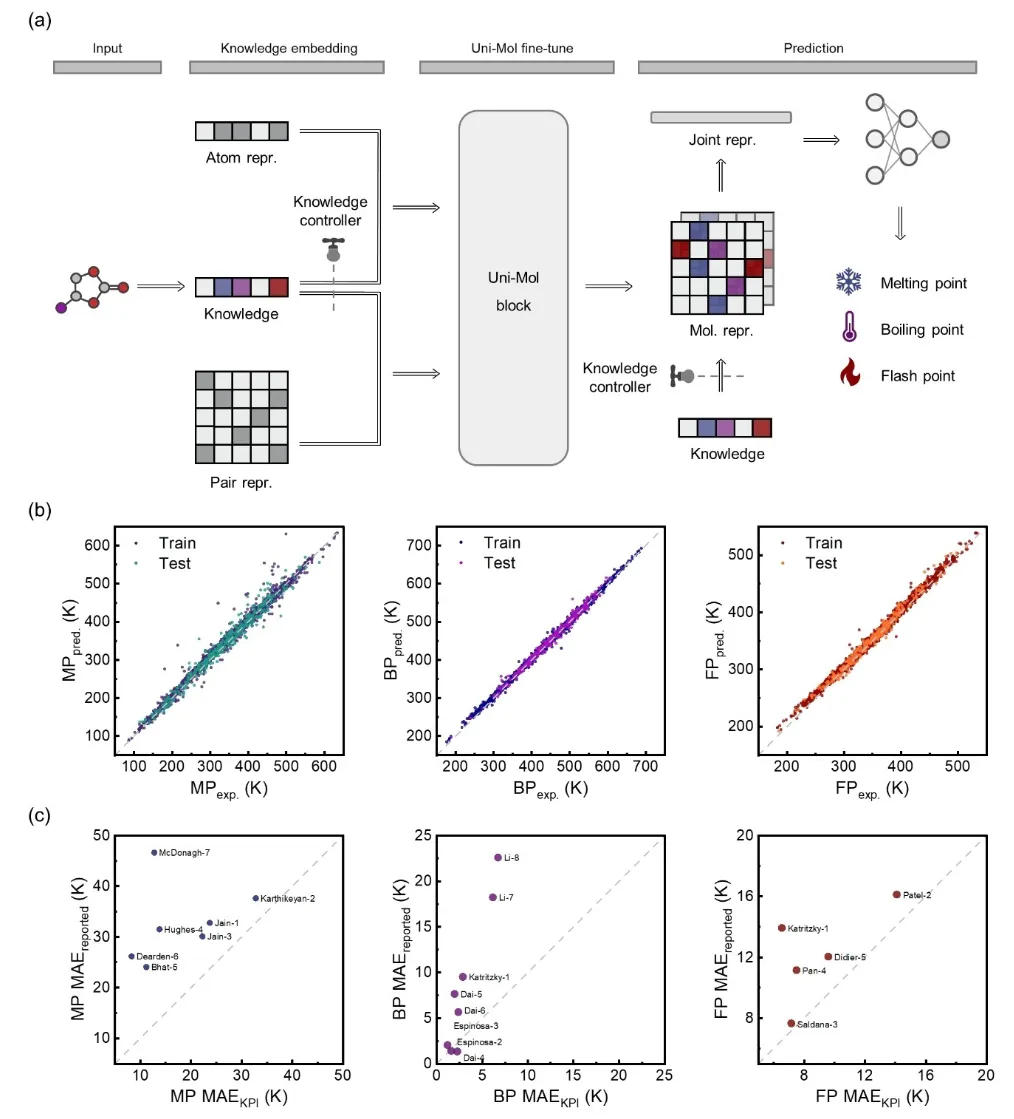
+**3.1 Overview of the KPI Framework Architecture**
+
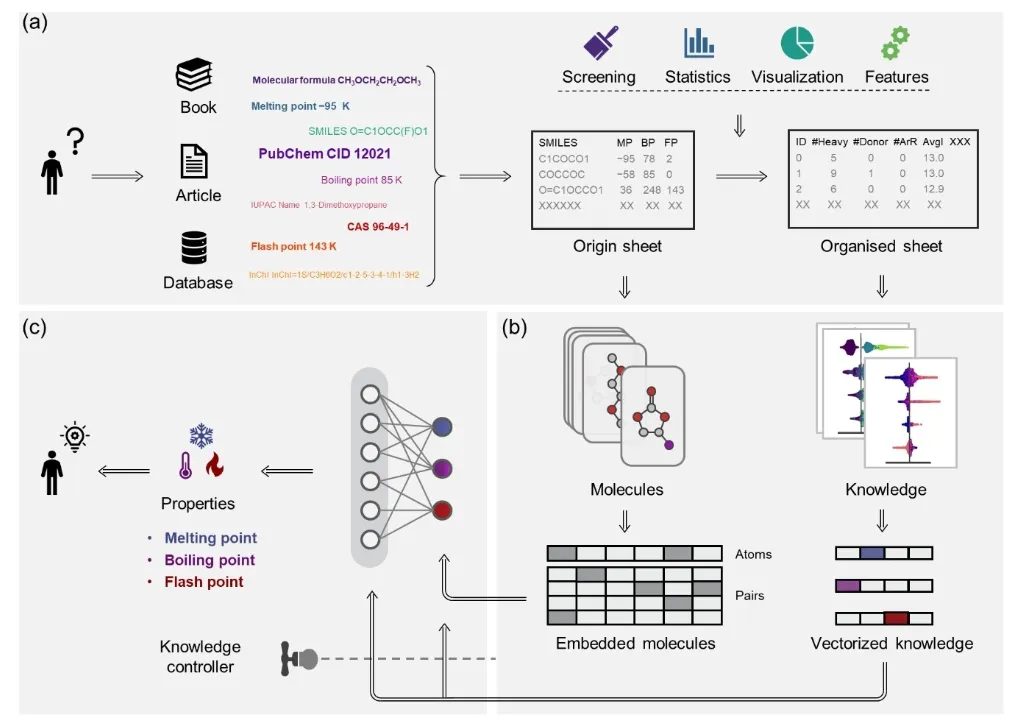
+Figure 1 provides a detailed illustration of the three core modules of the Knowledge–Data Dual-Driven Framework (KPI) and their collaborative operational process. First, the data organization and statistical analysis module collects molecular structure data along with properties such as melting point, boiling point, and flash point from public databases and literature. Through data cleaning and statistical analysis, the information is organized into structured tables. Clustering analysis and chemical space visualization techniques are then used for preliminary screening of potential molecules, laying the groundwork for subsequent research. Next, the interpretability and knowledge discovery module extracts molecular descriptors (e.g., the number of atoms, bond types, functional groups) and employs the Shapley Additive Explanations (SHAP) algorithm to reveal microscopic relationships between molecular structures and properties. The generated chemical knowledge serves as prior information for downstream models, enhancing the scientific interpretability and accuracy of predictions. In the knowledge-based molecular property prediction module, Uni-Mol is employed for molecular property prediction. Its pretrained model provides robust molecular representation capabilities, capturing complex structural features. By integrating chemical knowledge during fine-tuning, Uni-Mol embeds knowledge vectors at molecular, bond, and atomic levels. These vectors are dynamically optimized by the knowledge purity and knowledge flow controllers, achieving high-precision predictions for target properties such as melting point, boiling point, and flash point.
+
+
+
+### **Conclusion**
+
+DeepGlycanSite is a highly efficient prediction tool that leverages Uni-Mol’s robust molecular representation capabilities to enhance the accuracy of carbohydrate-binding site predictions on proteins. By integrating sequence and structural information, DeepGlycanSite not only surpasses traditional methods in detecting monosaccharide or disaccharide binding sites but also excels in identifying multiple binding sites. This provides critical insights into carbohydrate-protein interactions.
+
+Uni-Mol's ability to precisely capture chemical features and significantly improve predictive performance has established DeepGlycanSite as a powerful tool for addressing complex biological tasks. Its low dependence on protein structural accuracy enables analysis using predicted structures, supporting research into carbohydrate biological functions and drug development.
+
+The study encourages researchers to explore Uni-Mol for various downstream applications in different domains. The team welcomes collaboration and discussion to unlock further possibilities!
+
+Reference:
+[1] He, X., Zhao, L., Tian, Y. et al. Highly accurate carbohydrate-binding site prediction with DeepGlycanSite. Nat Commun 15, 5163 (2024). https://doi.org/10.1038/s41467-024-49516-2
+[2] Zhou G, Gao Z, Ding Q, Zheng H, Xu H, Wei Z, et al. Uni-Mol: A Universal 3D Molecular Representation Learning Framework. ChemRxiv. 2023; doi:10.26434/chemrxiv-2022-jjm0j-v4
\ No newline at end of file
diff --git a/source/_posts/unimol_kpi.md b/source/_posts/unimol_kpi.md
new file mode 100644
index 00000000..8770cc6a
--- /dev/null
+++ b/source/_posts/unimol_kpi.md
@@ -0,0 +1,84 @@
+---
+title: "What Can Uni-Mol Do too? | Predicting the molecular properties of battery electrolytes with the integrated KPI framework"
+date: 2024-12-02
+categories:
+- Uni-Mol
+mathjax: true
+---
+
+With the growing market demand for efficient and safe rechargeable batteries that can operate under extreme temperature conditions, the rapid and accurate evaluation of key properties of electrolyte molecules has become particularly important. A recent paper titled "A Knowledge–Data Dual-Driven Framework for Predicting the Molecular Properties of Rechargeable Battery Electrolytes," published in Angewandte Chemie International Edition, details an innovative approach known as the "Knowledge–Data Dual-Driven Framework" (KPI) specifically designed to predict the molecular properties of battery electrolytes, including melting point (MP), boiling point (BP), and flash point (FP). The research team skillfully combined deep learning techniques with domain-specific chemical knowledge, supported by large-scale datasets, significantly enhancing the accuracy and efficiency of predictions. In this framework, the Uni-Mol model plays a central role, demonstrating great potential in predicting the properties of electrolyte molecules and providing strong support for the development of next-generation high-performance batteries.
+
+Xiang Chen, an associate research fellow in the Department of Chemical Engineering at Tsinghua University, is the corresponding author of the paper. Yuchen Gao, a 2022 direct-entry PhD student in the Department of Chemical Engineering, is the first author. Co-authors include Yuhang Yuan, an undergraduate from the Tsinghua Academy of Wisdom; Suozhi Huang, an undergraduate from the Institute for Interdisciplinary Information Sciences; Nan Yao, a 2020 direct-entry PhD student; Legeng Yu, a 2021 direct-entry PhD student; Yaopeng Chen, a 2023 direct-entry PhD student; and Qiang Zhang, a professor in the Department of Chemical Engineering. The research was supported by funding from the National Natural Science Foundation of China, the National Key R&D Program of China, and the Beijing Natural Science Foundation.
+
+
+
+## 1. Research Background
+
+Secondary batteries have been widely used in electric vehicles and portable electronic devices. However, with the diversification of application scenarios—such as extreme cold, desert regions, and specialized settings like aerospace, underground exploration, and medical device sterilization—the demand for batteries to maintain stable performance under extreme temperatures has become increasingly urgent.
+
+In high-temperature environments, side reactions between electrolytes and active materials intensify, not only consuming electrolytes and active substances but also potentially triggering thermal runaway, posing serious safety risks. Conversely, in low-temperature conditions, the kinetics of electrochemical reactions are significantly reduced, which can lead to the formation of lithium dendrites, further increasing safety hazards. Therefore, developing batteries capable of safe operation across a wide temperature range has become a critical technical challenge.
+
+Moreover, battery performance heavily depends on the physicochemical properties of electrolytes, such as melting point, boiling point, and flash point. Traditional experimental methods, however, are inefficient due to limited data and reliance on trial-and-error processes, making them inadequate for the rapid development of new electrolyte molecules. Against this backdrop, efficient molecular property prediction models and high-throughput screening tools are particularly important to support the advancement of battery research toward higher performance and enhanced safety.
+
+## 2. Methods
+In this paper, the research team developed an innovative approach called the Knowledge–Data Dual-Driven Framework (KPI) for predicting the molecular properties of secondary battery electrolytes. The KPI framework consists of three main modules: data organization and statistical analysis, interpretability and knowledge discovery, and knowledge-based molecular property prediction. First, the KPI framework collects molecular structure and property data from public databases and literature and automatically organizes it into structured datasets. Next, it employs interpretable machine learning methods to explore structure–property relationships from a microscopic perspective. Finally, the discovered knowledge is embedded into property prediction models.
+
+Uni-Mol, serving as a foundational model, is utilized for molecular property prediction within the framework. It integrates a knowledge embedding mechanism to enhance prediction performance through pretraining and fine-tuning.
+
+1. **Pretraining**:
+ Uni-Mol undergoes pretraining on a large dataset of chemical molecules (in SMILES notation) to learn the underlying relationships between molecular structures and properties. This process provides a robust molecular representation foundation for property prediction.
+
+2. **Knowledge Embedding and Fine-Tuning**:
+ For specific tasks (e.g., predicting melting point, boiling point, and flash point), Uni-Mol integrates critical information extracted from data and chemical knowledge through a knowledge embedding mechanism. This mechanism uses a knowledge purity controller and a knowledge flow controller to adjust the proportion and dimensionality of embedded knowledge vectors, significantly improving the model's prediction accuracy.
+
+3. **Efficient Screening and Molecular Design (KPI-Assisted Electrolyte Molecular Design)**:
+ The fine-tuned Uni-Mol achieves high accuracy in predicting molecular properties, enabling high-throughput screening to quickly identify molecules with excellent performance. This supports the development of wide-temperature-range and high-safety electrolytes.
+
+## 3. Results and Discussion
+**3.1 Overview of the KPI Framework Architecture**
+
+Figure 1 provides a detailed illustration of the three core modules of the Knowledge–Data Dual-Driven Framework (KPI) and their collaborative operational process. First, the data organization and statistical analysis module collects molecular structure data along with properties such as melting point, boiling point, and flash point from public databases and literature. Through data cleaning and statistical analysis, the information is organized into structured tables. Clustering analysis and chemical space visualization techniques are then used for preliminary screening of potential molecules, laying the groundwork for subsequent research. Next, the interpretability and knowledge discovery module extracts molecular descriptors (e.g., the number of atoms, bond types, functional groups) and employs the Shapley Additive Explanations (SHAP) algorithm to reveal microscopic relationships between molecular structures and properties. The generated chemical knowledge serves as prior information for downstream models, enhancing the scientific interpretability and accuracy of predictions. In the knowledge-based molecular property prediction module, Uni-Mol is employed for molecular property prediction. Its pretrained model provides robust molecular representation capabilities, capturing complex structural features. By integrating chemical knowledge during fine-tuning, Uni-Mol embeds knowledge vectors at molecular, bond, and atomic levels. These vectors are dynamically optimized by the knowledge purity and knowledge flow controllers, achieving high-precision predictions for target properties such as melting point, boiling point, and flash point.
+ +
+---
+
+**3.2 Data Analysis and SHAP Visualization in the KPI Framework**
+
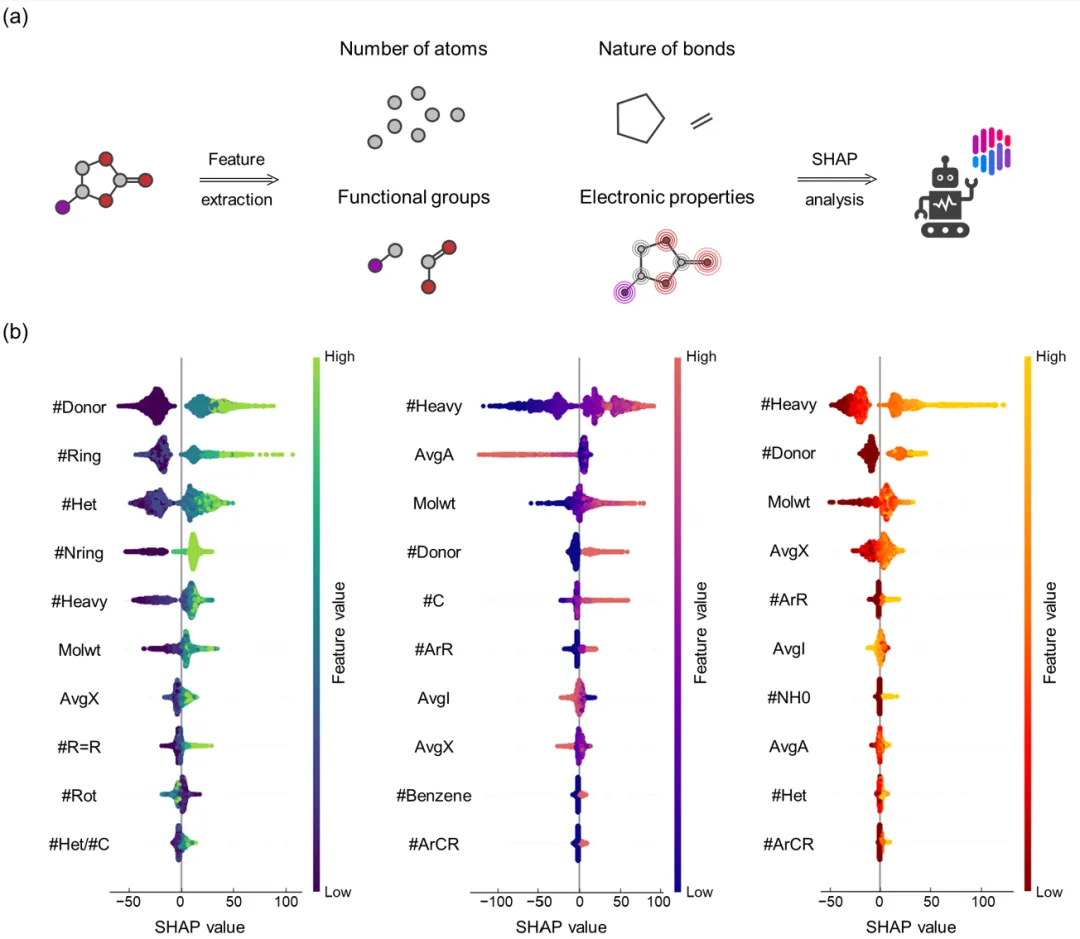
+Figure 2 uses the SHAP algorithm to analyze the contributions of molecular features to melting point, boiling point, and flash point, identifying the key factors influencing model decisions. The study extracted 64 molecular descriptors (e.g., number of atoms, bond properties, functional groups) and focused on the top ten important features for each property.
+
+For melting point, the key influencing factors include the number of hydrogen bond donors (#Donor), the number of rings (#Ring), and the number of double bonds (#R=R). Among these, hydrogen bond donors significantly increase melting points by enhancing intermolecular attractions. For boiling point and flash point, major features include the number of heavy atoms (#Heavy) and molecular weight (Molwt), reflecting the higher thermal stability of larger molecules. Additionally, electronic properties such as average electronegativity (AvgX) also moderately influence boiling point and flash point. These analyses uncover the deep correlations between molecular structures and properties, providing valuable references for model prediction and molecular design.
+
+
+---
+
+**3.2 Data Analysis and SHAP Visualization in the KPI Framework**
+
+Figure 2 uses the SHAP algorithm to analyze the contributions of molecular features to melting point, boiling point, and flash point, identifying the key factors influencing model decisions. The study extracted 64 molecular descriptors (e.g., number of atoms, bond properties, functional groups) and focused on the top ten important features for each property.
+
+For melting point, the key influencing factors include the number of hydrogen bond donors (#Donor), the number of rings (#Ring), and the number of double bonds (#R=R). Among these, hydrogen bond donors significantly increase melting points by enhancing intermolecular attractions. For boiling point and flash point, major features include the number of heavy atoms (#Heavy) and molecular weight (Molwt), reflecting the higher thermal stability of larger molecules. Additionally, electronic properties such as average electronegativity (AvgX) also moderately influence boiling point and flash point. These analyses uncover the deep correlations between molecular structures and properties, providing valuable references for model prediction and molecular design.
+ +
+---
+
+**3.3 Knowledge-Based Learning Models and Performance Evaluation**
+
+**1. Knowledge-Embedded Model Architecture**
+The model takes molecular conformations as input, encodes information about atoms and bonds, and uses Uni-Mol as the foundational model for molecular structure representation. Uni-Mol’s base molecular representation is adjusted using the knowledge purity and knowledge flow controllers, embedding multi-level chemical knowledge (including molecular, bond, and atomic levels) to enhance prediction performance. The knowledge-enhanced molecular representation is further optimized using a Transformer encoder to accurately predict target molecular properties such as melting point, boiling point, and flash point.
+
+**2. Prediction Results and Accuracy**
+The figures display the prediction results for melting point, boiling point, and flash point, with training and test sets distinguished by color. Data points closer to the diagonal line indicate higher prediction accuracy. The integration of Uni-Mol’s robust molecular representation capabilities with embedded knowledge significantly improves prediction precision.
+
+**3. Model Comparisons**
+Tests across various datasets demonstrate that the KPI framework achieves state-of-the-art (SOTA) performance on 18 benchmark datasets. Compared to the Uni-Mol model without knowledge embedding or traditional methods (e.g., Random Forest), the knowledge-embedded Uni-Mol significantly reduces prediction errors (MAE).
+
+
+
+---
+
+**3.3 Knowledge-Based Learning Models and Performance Evaluation**
+
+**1. Knowledge-Embedded Model Architecture**
+The model takes molecular conformations as input, encodes information about atoms and bonds, and uses Uni-Mol as the foundational model for molecular structure representation. Uni-Mol’s base molecular representation is adjusted using the knowledge purity and knowledge flow controllers, embedding multi-level chemical knowledge (including molecular, bond, and atomic levels) to enhance prediction performance. The knowledge-enhanced molecular representation is further optimized using a Transformer encoder to accurately predict target molecular properties such as melting point, boiling point, and flash point.
+
+**2. Prediction Results and Accuracy**
+The figures display the prediction results for melting point, boiling point, and flash point, with training and test sets distinguished by color. Data points closer to the diagonal line indicate higher prediction accuracy. The integration of Uni-Mol’s robust molecular representation capabilities with embedded knowledge significantly improves prediction precision.
+
+**3. Model Comparisons**
+Tests across various datasets demonstrate that the KPI framework achieves state-of-the-art (SOTA) performance on 18 benchmark datasets. Compared to the Uni-Mol model without knowledge embedding or traditional methods (e.g., Random Forest), the knowledge-embedded Uni-Mol significantly reduces prediction errors (MAE).
+
+ +
+---
+
+**3.4 Facilitating Advanced Molecular Screening and Design with the KPI Framework**
+
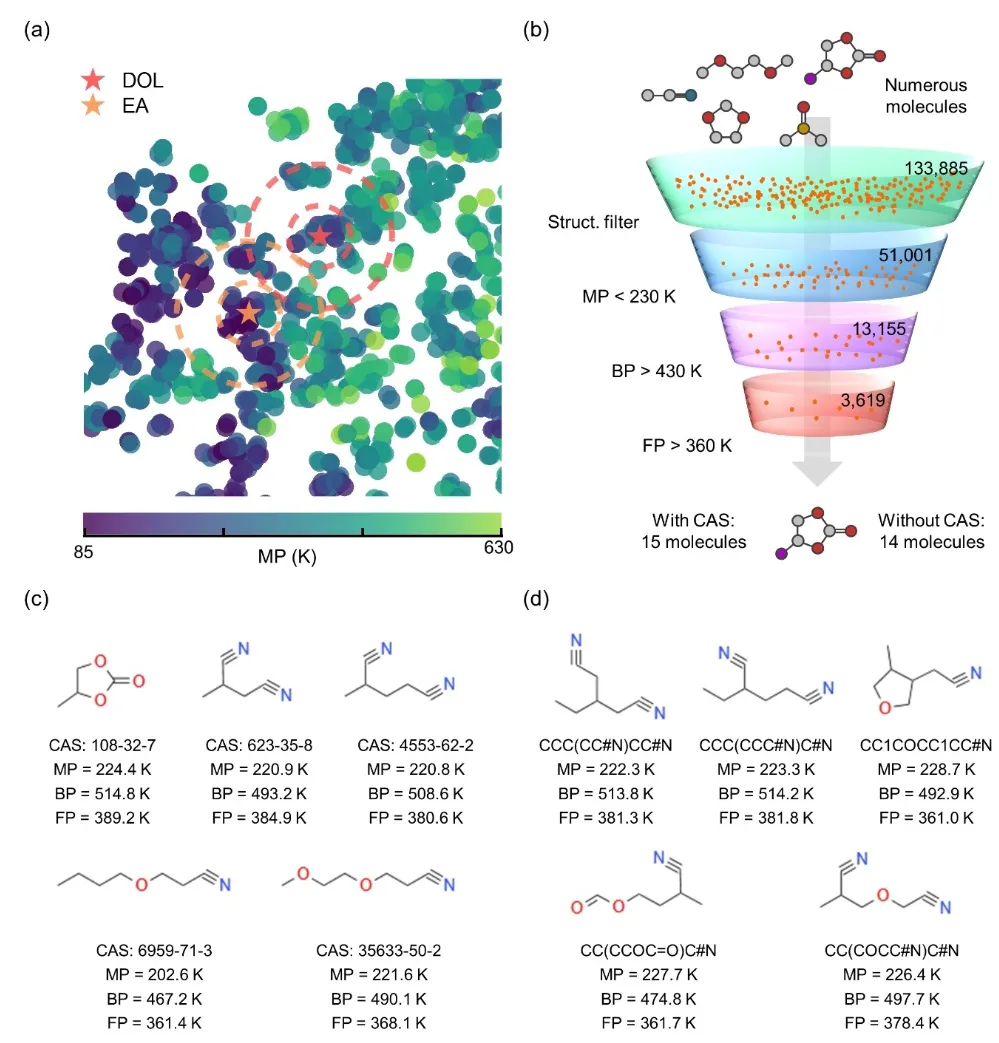
+Figure 4 showcases the specific applications of the KPI framework in molecular proximity search, high-throughput screening, and the presentation of screening results. In molecular proximity search, performance-optimized molecules (e.g., 1,3-dioxolane, DOL) serve as centers for first- and second-degree searches, identifying candidate molecules with similar properties, such as tetrahydrofuran (THF).
+
+For high-throughput screening, the KPI framework processes extensive databases (e.g., QM9 with 133,885 molecules) to identify molecules meeting criteria such as a melting point below 230 K, a boiling point above 430 K, and a flash point above 360 K. Multi-level filtering significantly narrows the search scope, ultimately pinpointing molecules with potential for wide-temperature-range and high-safety applications.
+
+In the presentation of screening results, Figure 4c lists 15 registered molecules (with CAS IDs), including their molecular structures, SMILES notations, and properties such as melting point, boiling point, and flash point. Figure 4d highlights 14 unregistered molecules (without CAS IDs), further demonstrating the KPI framework’s powerful capabilities in discovering novel molecules.
+
+
+---
+
+**3.4 Facilitating Advanced Molecular Screening and Design with the KPI Framework**
+
+Figure 4 showcases the specific applications of the KPI framework in molecular proximity search, high-throughput screening, and the presentation of screening results. In molecular proximity search, performance-optimized molecules (e.g., 1,3-dioxolane, DOL) serve as centers for first- and second-degree searches, identifying candidate molecules with similar properties, such as tetrahydrofuran (THF).
+
+For high-throughput screening, the KPI framework processes extensive databases (e.g., QM9 with 133,885 molecules) to identify molecules meeting criteria such as a melting point below 230 K, a boiling point above 430 K, and a flash point above 360 K. Multi-level filtering significantly narrows the search scope, ultimately pinpointing molecules with potential for wide-temperature-range and high-safety applications.
+
+In the presentation of screening results, Figure 4c lists 15 registered molecules (with CAS IDs), including their molecular structures, SMILES notations, and properties such as melting point, boiling point, and flash point. Figure 4d highlights 14 unregistered molecules (without CAS IDs), further demonstrating the KPI framework’s powerful capabilities in discovering novel molecules.
+ +
+## Conclusions
+This study proposes a dual-driven framework combining knowledge and data (KPI), offering a novel approach to the development of wide-temperature-range, high-safety electrolytes by accurately predicting the melting point, boiling point, and flash point of electrolyte molecules. The KPI framework not only achieves breakthroughs in predictive performance but also successfully identifies various potential molecules with outstanding properties through high-throughput screening and proximity searches.
+
+By leveraging the pretrained Uni-Mol model integrated with knowledge embedding techniques, the KPI framework significantly enhances the accuracy of molecular property predictions while providing a deeper understanding of the relationships between molecular structure and properties. The results demonstrate that this framework can greatly accelerate the design and discovery of novel electrolyte molecules, providing theoretical guidance and technical support for the development of next-generation batteries.
+
+In the future, the KPI framework is expected to find broad applications in other fields of molecular design, such as drug discovery and catalyst design, offering more efficient tools and methods to drive scientific innovation.
+
diff --git a/source/_posts/unimol_multi.md b/source/_posts/unimol_multi.md
new file mode 100644
index 00000000..69d46c8d
--- /dev/null
+++ b/source/_posts/unimol_multi.md
@@ -0,0 +1,109 @@
+---
+title: "What Can Uni-Mol Do too? | Multi-objective optimization unlocks a new evolutionary approach to chemical product design"
+date: 2024-10-16
+categories:
+- Uni-Mol
+mathjax: true
+---
+
+On July 15, 2024, Bilal Aslan from the University of Cape Town, Flavio Correa da Silva from the University of São Paulo, and Geoff Nitschke from the University of Cape Town collaborated to present their research titled “Multi-Objective Evolution for Chemical Product Design” at the Genetic and Evolutionary Computation Conference (GECCO). This study introduced a chemical product design method based on multi-objective evolutionary optimization. By innovatively integrating deep learning with evolutionary algorithms, the approach optimizes molecular properties and utilizes the Uni-Mol model to evaluate molecular toxicity, providing a novel solution for the design and optimization of chemical products.
+
+
+
+
+## Research Background
+In the field of chemical product design, optimizing target properties within molecular structures has always been a highly challenging task. Traditional optimization methods rely primarily on laboratory experiments, which are not only time-consuming and costly but also struggle to identify ideal molecular candidates within the vast design space of chemical molecules. With the advancement of computational technologies, methods based on deep learning and multi-objective evolutionary optimization have gradually emerged in chemical product design, enabling the rapid generation and screening of molecules that meet specific property requirements.
+
+However, these computational methods face challenges in comparing technical performance, as they must consider both quantitative evaluation and the diversity and innovation of generated molecular candidates. To address this, this study proposes a multi-objective evolutionary optimization framework that combines quantitative and qualitative evaluations. This approach aims to provide a more comprehensive comparison of different optimization techniques, facilitating more efficient molecular property optimization in chemical product design while ensuring that the generated molecules not only meet the required criteria but also exhibit innovation.
+
+## Uni-Mol Assists in Toxicity Assessment for Chemical Product Design
+
+In this study, Uni-Mol was employed to evaluate the aquatic toxicity of molecules in chemical product design. Uni-Mol is a universal 3D molecular representation learning framework, pretrained on the 3D structures of over 2.1 billion molecules. The research team utilized an RDKit-based pretrained 3D structural model and fine-tuned the Uni-Mol model using data extracted from the publicly available PubChem database, creating a model specifically designed for molecular toxicity assessment.
+
+During the molecule generation process, a multi-objective optimization method based on evolutionary algorithms was adopted. Initially, a set of seed molecules was selected from the PubChem database with a minimum similarity threshold of 80%. Uni-Mol was then used to evaluate molecular toxicity, excluding molecules with potential aquatic toxicity. The optimization process iteratively generated new molecules, selecting parent molecules based on the similarity threshold and generating offspring molecules according to hyperparameters (β and λ). The newly generated molecules were screened based on target properties, including minimizing molecular weight and complexity, maximizing XLogP, and ensuring non-toxicity. The optimization continued for multiple generations until reaching stability criteria or a runtime limit, ensuring that the resulting molecules met both functional and safety standards.
+
+This innovative application demonstrates the robust capability of Uni-Mol in molecular toxicity prediction. Figure 2 illustrates box plots of molecular properties such as complexity, molecular weight, XLogP, and reference similarity, all of which are linked to the optimization process (including toxicity assessment). The data indicate that these properties fluctuated toward target values during the optimization process, indirectly supporting Uni-Mol's role in molecular screening. This study highlights Uni-Mol's significant contribution to the safe design of chemical products.
+
+
+## Introduction to Multi-Objective Evolutionary Optimization (MOEO) Methods
+
+The implementation of multi-objective evolutionary optimization (MOEO) in chemical product design involves the following steps:
+
+1. **Optimization Objectives**:
+ The optimization aims to minimize molecular weight and complexity, ensure the optimal XLogP value, and eliminate molecules with aquatic toxicity.
+
+2. **Seed Molecule Selection**:
+ The process begins by selecting seed molecules with a similarity of at least 80% to reference molecules. The search space is defined around these seed molecules to ensure that candidate molecules retain characteristics similar to the seeds.
+
+3. **Toxicity Assessment**:
+ A Uni-Mol-trained model is used to evaluate the aquatic toxicity of molecules, filtering out those with potential toxicity to ensure safety.
+
+4. **Multi-Objective Optimization Process**:
+ Multi-objective optimization strategies, such as MO-CMA-ES, are applied to generate new candidate molecules based on the similarity to seed molecules. Parameters such as similarity thresholds, parent selection, and offspring selection are controlled to iteratively optimize the candidate set of molecules, ensuring their performance aligns with the defined objectives.
+
+
+
+## Conclusions
+This study proposes a dual-driven framework combining knowledge and data (KPI), offering a novel approach to the development of wide-temperature-range, high-safety electrolytes by accurately predicting the melting point, boiling point, and flash point of electrolyte molecules. The KPI framework not only achieves breakthroughs in predictive performance but also successfully identifies various potential molecules with outstanding properties through high-throughput screening and proximity searches.
+
+By leveraging the pretrained Uni-Mol model integrated with knowledge embedding techniques, the KPI framework significantly enhances the accuracy of molecular property predictions while providing a deeper understanding of the relationships between molecular structure and properties. The results demonstrate that this framework can greatly accelerate the design and discovery of novel electrolyte molecules, providing theoretical guidance and technical support for the development of next-generation batteries.
+
+In the future, the KPI framework is expected to find broad applications in other fields of molecular design, such as drug discovery and catalyst design, offering more efficient tools and methods to drive scientific innovation.
+
diff --git a/source/_posts/unimol_multi.md b/source/_posts/unimol_multi.md
new file mode 100644
index 00000000..69d46c8d
--- /dev/null
+++ b/source/_posts/unimol_multi.md
@@ -0,0 +1,109 @@
+---
+title: "What Can Uni-Mol Do too? | Multi-objective optimization unlocks a new evolutionary approach to chemical product design"
+date: 2024-10-16
+categories:
+- Uni-Mol
+mathjax: true
+---
+
+On July 15, 2024, Bilal Aslan from the University of Cape Town, Flavio Correa da Silva from the University of São Paulo, and Geoff Nitschke from the University of Cape Town collaborated to present their research titled “Multi-Objective Evolution for Chemical Product Design” at the Genetic and Evolutionary Computation Conference (GECCO). This study introduced a chemical product design method based on multi-objective evolutionary optimization. By innovatively integrating deep learning with evolutionary algorithms, the approach optimizes molecular properties and utilizes the Uni-Mol model to evaluate molecular toxicity, providing a novel solution for the design and optimization of chemical products.
+
+
+
+
+## Research Background
+In the field of chemical product design, optimizing target properties within molecular structures has always been a highly challenging task. Traditional optimization methods rely primarily on laboratory experiments, which are not only time-consuming and costly but also struggle to identify ideal molecular candidates within the vast design space of chemical molecules. With the advancement of computational technologies, methods based on deep learning and multi-objective evolutionary optimization have gradually emerged in chemical product design, enabling the rapid generation and screening of molecules that meet specific property requirements.
+
+However, these computational methods face challenges in comparing technical performance, as they must consider both quantitative evaluation and the diversity and innovation of generated molecular candidates. To address this, this study proposes a multi-objective evolutionary optimization framework that combines quantitative and qualitative evaluations. This approach aims to provide a more comprehensive comparison of different optimization techniques, facilitating more efficient molecular property optimization in chemical product design while ensuring that the generated molecules not only meet the required criteria but also exhibit innovation.
+
+## Uni-Mol Assists in Toxicity Assessment for Chemical Product Design
+
+In this study, Uni-Mol was employed to evaluate the aquatic toxicity of molecules in chemical product design. Uni-Mol is a universal 3D molecular representation learning framework, pretrained on the 3D structures of over 2.1 billion molecules. The research team utilized an RDKit-based pretrained 3D structural model and fine-tuned the Uni-Mol model using data extracted from the publicly available PubChem database, creating a model specifically designed for molecular toxicity assessment.
+
+During the molecule generation process, a multi-objective optimization method based on evolutionary algorithms was adopted. Initially, a set of seed molecules was selected from the PubChem database with a minimum similarity threshold of 80%. Uni-Mol was then used to evaluate molecular toxicity, excluding molecules with potential aquatic toxicity. The optimization process iteratively generated new molecules, selecting parent molecules based on the similarity threshold and generating offspring molecules according to hyperparameters (β and λ). The newly generated molecules were screened based on target properties, including minimizing molecular weight and complexity, maximizing XLogP, and ensuring non-toxicity. The optimization continued for multiple generations until reaching stability criteria or a runtime limit, ensuring that the resulting molecules met both functional and safety standards.
+
+This innovative application demonstrates the robust capability of Uni-Mol in molecular toxicity prediction. Figure 2 illustrates box plots of molecular properties such as complexity, molecular weight, XLogP, and reference similarity, all of which are linked to the optimization process (including toxicity assessment). The data indicate that these properties fluctuated toward target values during the optimization process, indirectly supporting Uni-Mol's role in molecular screening. This study highlights Uni-Mol's significant contribution to the safe design of chemical products.
+
+
+## Introduction to Multi-Objective Evolutionary Optimization (MOEO) Methods
+
+The implementation of multi-objective evolutionary optimization (MOEO) in chemical product design involves the following steps:
+
+1. **Optimization Objectives**:
+ The optimization aims to minimize molecular weight and complexity, ensure the optimal XLogP value, and eliminate molecules with aquatic toxicity.
+
+2. **Seed Molecule Selection**:
+ The process begins by selecting seed molecules with a similarity of at least 80% to reference molecules. The search space is defined around these seed molecules to ensure that candidate molecules retain characteristics similar to the seeds.
+
+3. **Toxicity Assessment**:
+ A Uni-Mol-trained model is used to evaluate the aquatic toxicity of molecules, filtering out those with potential toxicity to ensure safety.
+
+4. **Multi-Objective Optimization Process**:
+ Multi-objective optimization strategies, such as MO-CMA-ES, are applied to generate new candidate molecules based on the similarity to seed molecules. Parameters such as similarity thresholds, parent selection, and offspring selection are controlled to iteratively optimize the candidate set of molecules, ensuring their performance aligns with the defined objectives.
+
+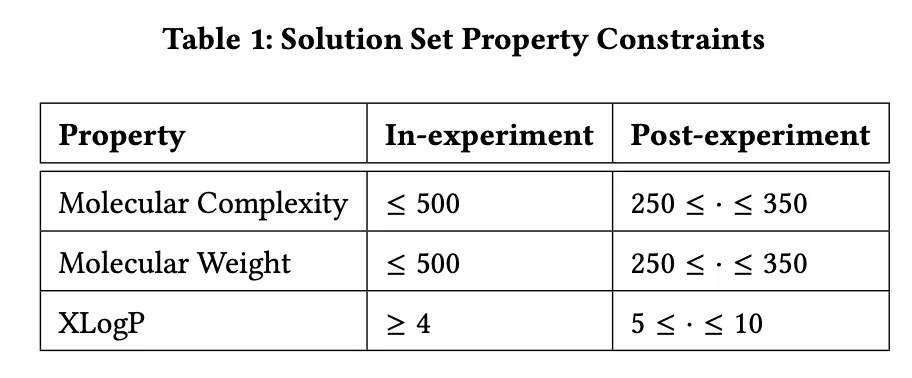 +
+**Table 1: Attribute Constraints for Chemical Product Design**
+
+Table 1 presents the attribute constraints used in the experiment for chemical product design. The table is divided into two parts: **In-experiment Constraints** and **Post-experiment Constraints**.
+
+1. **Molecular Complexity**:
+ - *In-experiment*: Molecular complexity is constrained to be ≤ 500.
+ - *Post-experiment*: Optimized molecular complexity must range between 250 and 350.
+
+2. **Molecular Weight**:
+ - *In-experiment*: Molecular weight is limited to ≤ 500.
+ - *Post-experiment*: Optimized molecular weight must range between 250 and 350.
+
+3. **XLogP (Lipophilicity Indicator)**:
+ - *In-experiment*: XLogP must be ≥ 4.
+ - *Post-experiment*: Optimized XLogP values must range between 5 and 10.
+
+These constraints are designed to ensure that the molecules generated during the optimization process exhibit both good performance and practical applicability. The in-experiment constraints are relatively lenient, allowing for broader exploration in the early stages. In contrast, the post-experiment constraints are stricter, ensuring that the final molecules meet specific standards for chemical product design.
+
+
+## Results and Discussion
+This section focuses on analyzing the effectiveness of applying the Multi-Objective Evolutionary Optimization (MOEO) method in chemical product design. The research team conducted 50 generations of evolutionary runs with 20 independent experiments to evaluate each method's performance in escaping local optima and promoting exploration. The results are summarized as follows:
+
+---
+
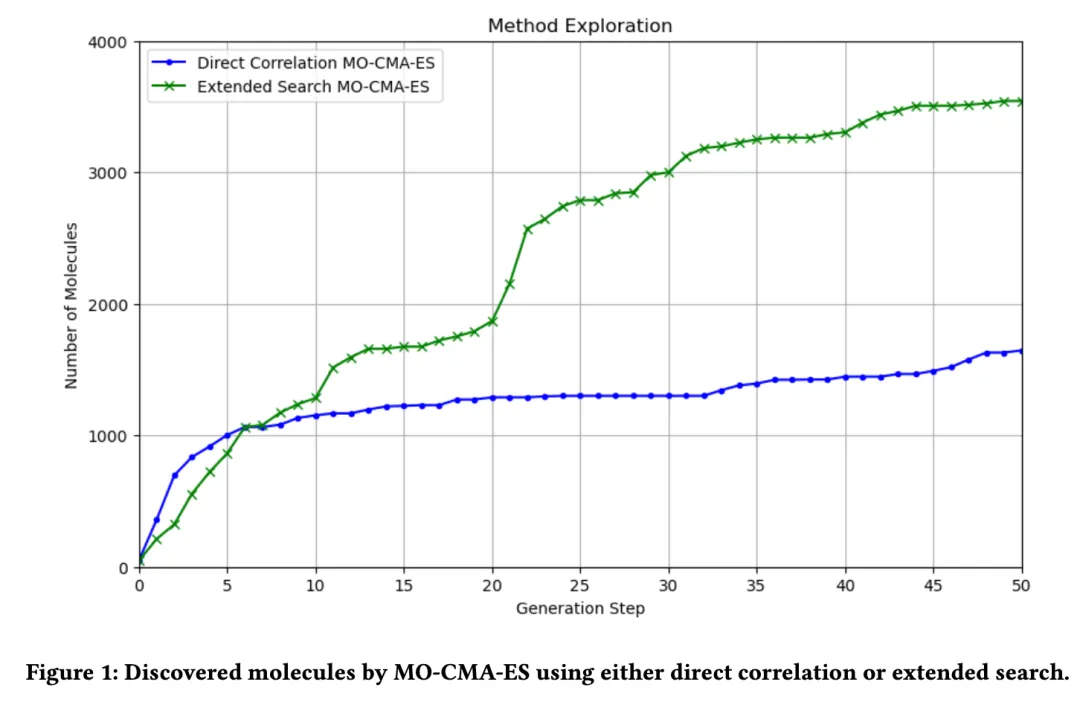
+**1. Diversity:**
+*Figure 1* illustrates the variation in the number of molecules generated during the evolutionary process using the MO-CMA-ES algorithm under two approaches: **directly related search** and **extended search**.
+
+- **Directly Related Search:**
+ This method generates fewer molecules, primarily searching around the initial seed molecules within a limited search space. As a result, molecular diversity is relatively low.
+
+- **Extended Search:**
+ Extended search allows the discovery of a larger number of molecular candidates, with the number of generated molecules gradually increasing throughout the evolutionary process. This demonstrates the method's advantage in expanding the search space and enhancing molecular diversity.
+
+
+**Table 1: Attribute Constraints for Chemical Product Design**
+
+Table 1 presents the attribute constraints used in the experiment for chemical product design. The table is divided into two parts: **In-experiment Constraints** and **Post-experiment Constraints**.
+
+1. **Molecular Complexity**:
+ - *In-experiment*: Molecular complexity is constrained to be ≤ 500.
+ - *Post-experiment*: Optimized molecular complexity must range between 250 and 350.
+
+2. **Molecular Weight**:
+ - *In-experiment*: Molecular weight is limited to ≤ 500.
+ - *Post-experiment*: Optimized molecular weight must range between 250 and 350.
+
+3. **XLogP (Lipophilicity Indicator)**:
+ - *In-experiment*: XLogP must be ≥ 4.
+ - *Post-experiment*: Optimized XLogP values must range between 5 and 10.
+
+These constraints are designed to ensure that the molecules generated during the optimization process exhibit both good performance and practical applicability. The in-experiment constraints are relatively lenient, allowing for broader exploration in the early stages. In contrast, the post-experiment constraints are stricter, ensuring that the final molecules meet specific standards for chemical product design.
+
+
+## Results and Discussion
+This section focuses on analyzing the effectiveness of applying the Multi-Objective Evolutionary Optimization (MOEO) method in chemical product design. The research team conducted 50 generations of evolutionary runs with 20 independent experiments to evaluate each method's performance in escaping local optima and promoting exploration. The results are summarized as follows:
+
+---
+
+**1. Diversity:**
+*Figure 1* illustrates the variation in the number of molecules generated during the evolutionary process using the MO-CMA-ES algorithm under two approaches: **directly related search** and **extended search**.
+
+- **Directly Related Search:**
+ This method generates fewer molecules, primarily searching around the initial seed molecules within a limited search space. As a result, molecular diversity is relatively low.
+
+- **Extended Search:**
+ Extended search allows the discovery of a larger number of molecular candidates, with the number of generated molecules gradually increasing throughout the evolutionary process. This demonstrates the method's advantage in expanding the search space and enhancing molecular diversity.
+ +
+---
+
+**2. Quality of Optimized Solution Sets:**
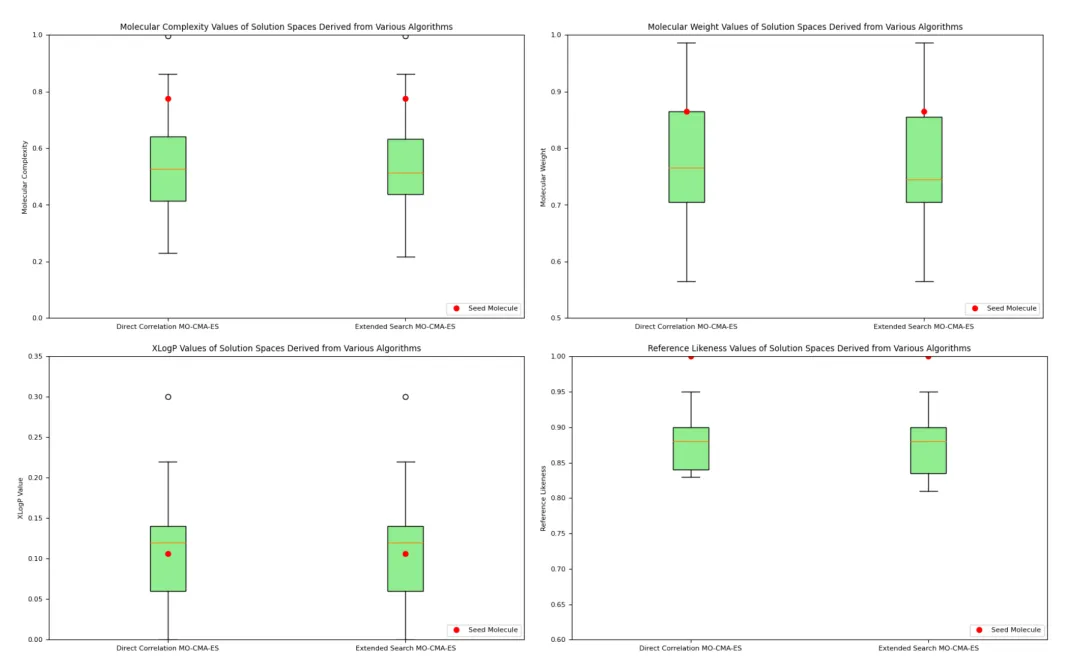
+*Figure 2* shows that after optimization using MO-CMA-ES, the properties of different molecules were well-controlled. While some properties (e.g., XLogP) exhibited significant fluctuations, the overall performance of the optimized molecules showed minimal variation within the target range, highlighting the effectiveness of the optimization method in maintaining both molecular characteristics and diversity.
+
+- **Top Left: Molecular Complexity**
+ This plot shows the distribution of optimized molecular complexity. The low height of the box plot indicates that the molecular complexity of the optimized molecules was concentrated within a narrow range, demonstrating the effectiveness of multi-objective optimization in reducing molecular complexity.
+
+- **Top Right: Molecular Weight**
+ The distribution of molecular weight was similarly optimized to a narrow range, indicating that the optimization method effectively controlled molecular weight to meet the expected standards.
+
+- **Bottom Left: XLogP (Lipophilicity)**
+ The XLogP values showed relatively large fluctuations but remained within the target range. The wider distribution indicates that more molecular candidates were explored during the optimization process.
+
+- **Bottom Right: Reference Similarity**
+ Reference similarity measures the similarity between optimized molecules and seed molecules. The results indicate high similarity for most generated molecules, showing that the key characteristics of the reference molecules were retained.
+
+
+
+---
+
+**2. Quality of Optimized Solution Sets:**
+*Figure 2* shows that after optimization using MO-CMA-ES, the properties of different molecules were well-controlled. While some properties (e.g., XLogP) exhibited significant fluctuations, the overall performance of the optimized molecules showed minimal variation within the target range, highlighting the effectiveness of the optimization method in maintaining both molecular characteristics and diversity.
+
+- **Top Left: Molecular Complexity**
+ This plot shows the distribution of optimized molecular complexity. The low height of the box plot indicates that the molecular complexity of the optimized molecules was concentrated within a narrow range, demonstrating the effectiveness of multi-objective optimization in reducing molecular complexity.
+
+- **Top Right: Molecular Weight**
+ The distribution of molecular weight was similarly optimized to a narrow range, indicating that the optimization method effectively controlled molecular weight to meet the expected standards.
+
+- **Bottom Left: XLogP (Lipophilicity)**
+ The XLogP values showed relatively large fluctuations but remained within the target range. The wider distribution indicates that more molecular candidates were explored during the optimization process.
+
+- **Bottom Right: Reference Similarity**
+ Reference similarity measures the similarity between optimized molecules and seed molecules. The results indicate high similarity for most generated molecules, showing that the key characteristics of the reference molecules were retained.
+
+ +
+---
+
+These results highlight the MOEO method's ability to balance the trade-off between molecular diversity and optimized property control, effectively supporting the chemical product design process.
+
+
+## Conclusions
+This work compares two heuristic search methods for multi-objective optimization (MOO) applied to chemical product design: **directly related search** and **extended search**. The experimental results demonstrate that extended search improves the diversity of solution sets without compromising solution quality. The primary contribution of this study lies in the development of appropriate evaluation metrics to compare different optimization strategies, particularly by assessing solution set diversity and quality.
+
+Uni-Mol played a critical role in the study, especially in molecular toxicity prediction. By leveraging the trained Uni-Mol model, the research effectively filtered out molecules with aquatic toxicity, ensuring that the optimization results not only achieved performance breakthroughs but also met safety standards.
+
+The results further revealed that the chemical design space is unevenly distributed in terms of molecular similarity and corresponding observable property values. This unevenness, particularly in multi-objective optimization scenarios, often leads to entrapment in local optima. Future work will focus on enhancing dynamic parameter optimization, fine-tuning parameter interactions (such as 𝛽 and 𝜆), and expanding the dataset using generative adversarial networks (GANs) and evolutionary transfer learning. These approaches aim to avoid local optima and generate more diverse solutions.
diff --git a/source/_posts/unimol_nag2g.md b/source/_posts/unimol_nag2g.md
new file mode 100644
index 00000000..cc875d2e
--- /dev/null
+++ b/source/_posts/unimol_nag2g.md
@@ -0,0 +1,194 @@
+---
+title: "What Can Uni-Mol Do too? | Unveiling NAG2G: A Powerful Tool for Enhancing Single-Step Retrosynthesis Prediction"
+date: 2024-08-08
+categories:
+- Uni-Mol
+mathjax: true
+---
+
+On February 13, 2024, DP Technology published a cover article in JACS Au titled "Node-Aligned Graph-to-Graph: Elevating Template-free Deep Learning Approaches in Single-Step Retrosynthesis." This study developed a Transformer-based Node-Aligned Graph-to-Graph (NAG2G) model, significantly improving the accuracy of single-step retrosynthesis prediction.
+
+The NAG2G model integrates 2D molecular graph and 3D conformation information, achieving atom mapping between products and reactants through node alignment. This approach overcomes the limitations of traditional template-based methods.
+
+This groundbreaking achievement provides a powerful tool for chemical synthesis design, advancing the field of retrosynthesis and setting a new standard for single-step prediction methodologies.
+
+
+
+
+## Research Background
+
+Single-step retrosynthesis (SSR) is a critical step in organic chemistry that involves reverse reasoning to synthesize a target product or intermediate. SSR plays a pivotal role in automated multi-step synthesis route design. With the advancement of computer-aided synthesis planning tools, researchers have turned to deep learning (DL) techniques to address this task.
+
+DL frameworks for SSR can be broadly categorized into three types: **template-based**, **semi-template-based**, and **template-free**.
+- **Template-based methods** rely on existing reaction rules for retrosynthesis design.
+- **Semi-template-based methods** involve a two-step process: identifying the reaction center and then predicting reactants.
+- **Template-free methods**, on the other hand, directly predict reactants using deep learning models.
+
+However, template-free methods often overlook critical 2D molecular information and struggle with atom alignment during node generation, leading to performance limitations compared to template-based and semi-template-based approaches.
+
+To overcome these challenges, this work introduces the **Node-Aligned Graph-to-Graph (NAG2G)** model, a Transformer-based template-free DL model. NAG2G integrates 2D molecular graph and 3D conformation information, preserving comprehensive molecular details. It employs node alignment to achieve atom mapping between products and reactants, generating the node graph step-by-step in an autoregressive manner.
+
+Through extensive benchmarking and case studies, this work demonstrates the exceptional performance of NAG2G in terms of prediction accuracy and practical applicability.
+
+## Methods
+### **Methodology**
+
+#### **2.1 Computer-Aided Single-Step Retrosynthesis (SSR) Workflow**
+
+*Figure 1* outlines the three primary design methods for computer-aided SSR: template-based, semi-template-based, and template-free approaches. Each has its own characteristics and limitations:
+
+1. **Template-Based Methods**:
+ These methods rely on a dictionary of known reaction rules, predicting retrosynthesis pathways by matching reaction templates. While intuitive, their coverage is limited to the template library, making it difficult to predict novel reactions outside the template scope.
+
+2. **Semi-Template-Based Methods**:
+ These divide the SSR task into two stages: first detecting synthons or intermediates, and then predicting the reactants. While this expands the search capacity to some extent, errors in each stage can accumulate, negatively impacting the final prediction results.
+
+3. **Template-Free Methods**:
+ Models like NAG2G use deep learning to directly predict reactants without relying on predefined templates. NAG2G integrates 2D molecular graphs and 3D conformations, leveraging node alignment to map atoms between products and reactants, significantly improving prediction accuracy.
+
+The workflow highlights NAG2G's innovative and advantageous ability to retain molecular detail and enhance prediction accuracy, overcoming the limitations of traditional template-based methods. This approach provides a flexible and efficient solution, revolutionizing chemical synthesis design tasks.
+
+
+
+---
+
+These results highlight the MOEO method's ability to balance the trade-off between molecular diversity and optimized property control, effectively supporting the chemical product design process.
+
+
+## Conclusions
+This work compares two heuristic search methods for multi-objective optimization (MOO) applied to chemical product design: **directly related search** and **extended search**. The experimental results demonstrate that extended search improves the diversity of solution sets without compromising solution quality. The primary contribution of this study lies in the development of appropriate evaluation metrics to compare different optimization strategies, particularly by assessing solution set diversity and quality.
+
+Uni-Mol played a critical role in the study, especially in molecular toxicity prediction. By leveraging the trained Uni-Mol model, the research effectively filtered out molecules with aquatic toxicity, ensuring that the optimization results not only achieved performance breakthroughs but also met safety standards.
+
+The results further revealed that the chemical design space is unevenly distributed in terms of molecular similarity and corresponding observable property values. This unevenness, particularly in multi-objective optimization scenarios, often leads to entrapment in local optima. Future work will focus on enhancing dynamic parameter optimization, fine-tuning parameter interactions (such as 𝛽 and 𝜆), and expanding the dataset using generative adversarial networks (GANs) and evolutionary transfer learning. These approaches aim to avoid local optima and generate more diverse solutions.
diff --git a/source/_posts/unimol_nag2g.md b/source/_posts/unimol_nag2g.md
new file mode 100644
index 00000000..cc875d2e
--- /dev/null
+++ b/source/_posts/unimol_nag2g.md
@@ -0,0 +1,194 @@
+---
+title: "What Can Uni-Mol Do too? | Unveiling NAG2G: A Powerful Tool for Enhancing Single-Step Retrosynthesis Prediction"
+date: 2024-08-08
+categories:
+- Uni-Mol
+mathjax: true
+---
+
+On February 13, 2024, DP Technology published a cover article in JACS Au titled "Node-Aligned Graph-to-Graph: Elevating Template-free Deep Learning Approaches in Single-Step Retrosynthesis." This study developed a Transformer-based Node-Aligned Graph-to-Graph (NAG2G) model, significantly improving the accuracy of single-step retrosynthesis prediction.
+
+The NAG2G model integrates 2D molecular graph and 3D conformation information, achieving atom mapping between products and reactants through node alignment. This approach overcomes the limitations of traditional template-based methods.
+
+This groundbreaking achievement provides a powerful tool for chemical synthesis design, advancing the field of retrosynthesis and setting a new standard for single-step prediction methodologies.
+
+
+
+
+## Research Background
+
+Single-step retrosynthesis (SSR) is a critical step in organic chemistry that involves reverse reasoning to synthesize a target product or intermediate. SSR plays a pivotal role in automated multi-step synthesis route design. With the advancement of computer-aided synthesis planning tools, researchers have turned to deep learning (DL) techniques to address this task.
+
+DL frameworks for SSR can be broadly categorized into three types: **template-based**, **semi-template-based**, and **template-free**.
+- **Template-based methods** rely on existing reaction rules for retrosynthesis design.
+- **Semi-template-based methods** involve a two-step process: identifying the reaction center and then predicting reactants.
+- **Template-free methods**, on the other hand, directly predict reactants using deep learning models.
+
+However, template-free methods often overlook critical 2D molecular information and struggle with atom alignment during node generation, leading to performance limitations compared to template-based and semi-template-based approaches.
+
+To overcome these challenges, this work introduces the **Node-Aligned Graph-to-Graph (NAG2G)** model, a Transformer-based template-free DL model. NAG2G integrates 2D molecular graph and 3D conformation information, preserving comprehensive molecular details. It employs node alignment to achieve atom mapping between products and reactants, generating the node graph step-by-step in an autoregressive manner.
+
+Through extensive benchmarking and case studies, this work demonstrates the exceptional performance of NAG2G in terms of prediction accuracy and practical applicability.
+
+## Methods
+### **Methodology**
+
+#### **2.1 Computer-Aided Single-Step Retrosynthesis (SSR) Workflow**
+
+*Figure 1* outlines the three primary design methods for computer-aided SSR: template-based, semi-template-based, and template-free approaches. Each has its own characteristics and limitations:
+
+1. **Template-Based Methods**:
+ These methods rely on a dictionary of known reaction rules, predicting retrosynthesis pathways by matching reaction templates. While intuitive, their coverage is limited to the template library, making it difficult to predict novel reactions outside the template scope.
+
+2. **Semi-Template-Based Methods**:
+ These divide the SSR task into two stages: first detecting synthons or intermediates, and then predicting the reactants. While this expands the search capacity to some extent, errors in each stage can accumulate, negatively impacting the final prediction results.
+
+3. **Template-Free Methods**:
+ Models like NAG2G use deep learning to directly predict reactants without relying on predefined templates. NAG2G integrates 2D molecular graphs and 3D conformations, leveraging node alignment to map atoms between products and reactants, significantly improving prediction accuracy.
+
+The workflow highlights NAG2G's innovative and advantageous ability to retain molecular detail and enhance prediction accuracy, overcoming the limitations of traditional template-based methods. This approach provides a flexible and efficient solution, revolutionizing chemical synthesis design tasks.
+
+ +
+---
+
+#### **2.2 The Role of Uni-Mol in NAG2G**
+
+Uni-Mol plays a critical role in processing and representing molecular information within the NAG2G model. By combining 2D molecular graphs and 3D conformations, NAG2G retains comprehensive molecular details, with Uni-Mol serving as a key enabler. Specifically, Uni-Mol's encoder transforms the 2D and 3D molecular information into compact numerical representations, allowing the model to better understand and process molecular structures.
+
+The NAG2G encoder, inspired by Uni-Mol, adopts a Transformer architecture to integrate 2D and 3D information. At each timestep, the encoder processes atom features, edge features, and 3D conformation coordinates to generate compact molecular representations. These representations, through node alignment, enable the model to accurately map atom relationships between products and reactants during the graph generation process.
+
+
+
+
+---
+
+#### **2.2 The Role of Uni-Mol in NAG2G**
+
+Uni-Mol plays a critical role in processing and representing molecular information within the NAG2G model. By combining 2D molecular graphs and 3D conformations, NAG2G retains comprehensive molecular details, with Uni-Mol serving as a key enabler. Specifically, Uni-Mol's encoder transforms the 2D and 3D molecular information into compact numerical representations, allowing the model to better understand and process molecular structures.
+
+The NAG2G encoder, inspired by Uni-Mol, adopts a Transformer architecture to integrate 2D and 3D information. At each timestep, the encoder processes atom features, edge features, and 3D conformation coordinates to generate compact molecular representations. These representations, through node alignment, enable the model to accurately map atom relationships between products and reactants during the graph generation process.
+
+
+ +
+---
+
+#### **2.3 Data Augmentation and Node Alignment**
+
+*Figure 3* illustrates the processes of data augmentation and product-reactant alignment:
+
+1. **Data Augmentation**:
+ Product SMILES sequences are randomly generated using RDKit, and input graph node orders are determined accordingly. This randomization increases input diversity, improving the model's robustness.
+
+2. **Node Alignment**:
+ Once the product graph node order is determined, reactant graph node orders are generated following a clear and unique rule:
+ - The order of shared atoms in the reactants is aligned with the product atom order.
+ - The order of non-shared atoms is extracted based on the aligned reactant SMILES sequence, ensuring a unique and consistent node order for reactants.
+
+
+
+
+---
+
+#### **2.3 Data Augmentation and Node Alignment**
+
+*Figure 3* illustrates the processes of data augmentation and product-reactant alignment:
+
+1. **Data Augmentation**:
+ Product SMILES sequences are randomly generated using RDKit, and input graph node orders are determined accordingly. This randomization increases input diversity, improving the model's robustness.
+
+2. **Node Alignment**:
+ Once the product graph node order is determined, reactant graph node orders are generated following a clear and unique rule:
+ - The order of shared atoms in the reactants is aligned with the product atom order.
+ - The order of non-shared atoms is extracted based on the aligned reactant SMILES sequence, ensuring a unique and consistent node order for reactants.
+
+
+ +
+---
+
+#### **2.4 Example Workflow of NAG2G Model**
+
+*Figure 4* demonstrates an example of the NAG2G model's generation process, highlighting how it predicts reactant molecular graph structures from products using node alignment and stepwise graph generation.
+
+1. **Initial Input**:
+ The model takes the product's molecular graph as input, including atom, edge, and 3D conformation information.
+
+2. **Node Generation**:
+ At each timestep, the decoder generates a new node based on the current molecular graph structure and previously generated nodes. The node alignment strategy ensures the generated node sequence aligns with the product's node order.
+
+3. **Edge Generation**:
+ Along with node generation, the decoder predicts edges connecting new nodes to existing nodes, determining bond types and relationships.
+
+4. **Stepwise Reactant Graph Construction**:
+ By repeating the above steps, the model iteratively constructs the complete reactant molecular graph while ensuring accurate alignment with the product graph.
+
+
+
+
+---
+
+#### **2.4 Example Workflow of NAG2G Model**
+
+*Figure 4* demonstrates an example of the NAG2G model's generation process, highlighting how it predicts reactant molecular graph structures from products using node alignment and stepwise graph generation.
+
+1. **Initial Input**:
+ The model takes the product's molecular graph as input, including atom, edge, and 3D conformation information.
+
+2. **Node Generation**:
+ At each timestep, the decoder generates a new node based on the current molecular graph structure and previously generated nodes. The node alignment strategy ensures the generated node sequence aligns with the product's node order.
+
+3. **Edge Generation**:
+ Along with node generation, the decoder predicts edges connecting new nodes to existing nodes, determining bond types and relationships.
+
+4. **Stepwise Reactant Graph Construction**:
+ By repeating the above steps, the model iteratively constructs the complete reactant molecular graph while ensuring accurate alignment with the product graph.
+
+
+ +
+---
+
+#### **2.5 Decoder Attention Mechanism: Enhancing NAG2G Generation Efficiency**
+
+The decoder generates reactant molecular graphs autoregressively. *Figure 5* explains how the attention mechanism efficiently handles time-varying graph features at each timestep:
+
+1. **Autoregressive Generation**:
+ At each timestep, the decoder generates a new node based on previously generated nodes, edges, and current graph features. To ensure accuracy, the decoder uses the true output from the previous step as input for the current step.
+
+2. **Multi-Head Attention**:
+ Multi-Head Attention is employed to process the query, key, and value matrices in parallel at each timestep. The computational complexity of each attention head is \( n \times n \times d_h \), with peak memory consumption of \( n \times n \times n \).
+
+3. **Time-Varying Graph Features**:
+ A time-varying graph feature matrix \( D \) is introduced into the attention mechanism as additive positional encoding. This ensures that graph features at each timestep are correctly utilized during the generation process.
+
+4. **Memory Optimization**:
+ To reduce computational costs and memory consumption, *Figure 5* illustrates an optimization method that reduces the dimension of matrix \( D \), lowering peak memory usage to \( n \times n \times d_h^2 \). This makes the attention mechanism more efficient for real-world applications.
+
+The decoder's attention mechanism ensures efficient and accurate graph generation, leveraging dynamic graph features at each timestep. This design enables the model to capture temporal dependencies during reactant graph construction, achieving high-precision single-step retrosynthesis prediction.
+
+
+
+
+---
+
+#### **2.5 Decoder Attention Mechanism: Enhancing NAG2G Generation Efficiency**
+
+The decoder generates reactant molecular graphs autoregressively. *Figure 5* explains how the attention mechanism efficiently handles time-varying graph features at each timestep:
+
+1. **Autoregressive Generation**:
+ At each timestep, the decoder generates a new node based on previously generated nodes, edges, and current graph features. To ensure accuracy, the decoder uses the true output from the previous step as input for the current step.
+
+2. **Multi-Head Attention**:
+ Multi-Head Attention is employed to process the query, key, and value matrices in parallel at each timestep. The computational complexity of each attention head is \( n \times n \times d_h \), with peak memory consumption of \( n \times n \times n \).
+
+3. **Time-Varying Graph Features**:
+ A time-varying graph feature matrix \( D \) is introduced into the attention mechanism as additive positional encoding. This ensures that graph features at each timestep are correctly utilized during the generation process.
+
+4. **Memory Optimization**:
+ To reduce computational costs and memory consumption, *Figure 5* illustrates an optimization method that reduces the dimension of matrix \( D \), lowering peak memory usage to \( n \times n \times d_h^2 \). This makes the attention mechanism more efficient for real-world applications.
+
+The decoder's attention mechanism ensures efficient and accurate graph generation, leveraging dynamic graph features at each timestep. This design enables the model to capture temporal dependencies during reactant graph construction, achieving high-precision single-step retrosynthesis prediction.
+
+
+ +
+## 3.Results and Discussion
+### **3.1 Model Evaluation**
+
+#### **Performance on USPTO-50k Dataset**
+
+
+
+## 3.Results and Discussion
+### **3.1 Model Evaluation**
+
+#### **Performance on USPTO-50k Dataset**
+
+ +
+*Table 1* presents the Top-k accuracy of the NAG2G model on the USPTO-50k dataset, compared against other existing models, including template-based, semi-template-based, and template-free approaches. Results show that NAG2G outperforms all other models across all evaluation metrics, particularly excelling in Top-1 and Top-10 accuracy.
+
+
+
+#### **Performance on USPTO-Full Dataset**
+
+
+
+*Table 1* presents the Top-k accuracy of the NAG2G model on the USPTO-50k dataset, compared against other existing models, including template-based, semi-template-based, and template-free approaches. Results show that NAG2G outperforms all other models across all evaluation metrics, particularly excelling in Top-1 and Top-10 accuracy.
+
+
+
+#### **Performance on USPTO-Full Dataset**
+
+ +
+*Table 2* highlights the Top-k accuracy of NAG2G on the larger-scale USPTO-Full dataset. Despite the increased task complexity compared to USPTO-50k, NAG2G consistently demonstrates strong predictive capabilities, outperforming most existing methods.
+
+
+
+
+#### **Ablation Study**
+
+
+
+*Table 2* highlights the Top-k accuracy of NAG2G on the larger-scale USPTO-Full dataset. Despite the increased task complexity compared to USPTO-50k, NAG2G consistently demonstrates strong predictive capabilities, outperforming most existing methods.
+
+
+
+
+#### **Ablation Study**
+
+ +
+*Table 3* presents an ablation analysis that explores the importance of various components in the NAG2G model, such as node alignment, data augmentation, and graph features. By systematically removing these components, the study evaluates their impact on model performance. Results indicate that **node alignment** and **data augmentation** are crucial for improving prediction accuracy.
+
+
+
+#### **Molecular Validity**
+
+
+
+*Table 3* presents an ablation analysis that explores the importance of various components in the NAG2G model, such as node alignment, data augmentation, and graph features. By systematically removing these components, the study evaluates their impact on model performance. Results indicate that **node alignment** and **data augmentation** are crucial for improving prediction accuracy.
+
+
+
+#### **Molecular Validity**
+
+ +
+*Table 4* evaluates the Top-k validity of molecules generated by NAG2G on the USPTO-50k dataset. The results demonstrate NAG2G's exceptional performance in generating valid molecules, further verifying its reliability in molecular generation tasks.
+
+---
+
+### **3.2 Practical Application Cases: Exceptional Performance of NAG2G in Drug Molecule Retrosynthesis Prediction**
+
+*Figure 6* showcases the retrosynthesis pathways for four drug molecules (**Nirmatrelvir**, **Osimertinib**, **Salmeterol**, and **Lenalidomide**) and provides detailed examples of NAG2G’s performance in predicting these pathways. These cases validate the accuracy and efficacy of NAG2G in real-world applications.
+
+1. **Nirmatrelvir**:
+ - The figure displays a six-step synthesis pathway for Nirmatrelvir.
+ - NAG2G successfully predicted all reaction steps, with the top-ranked predictions for the first three steps, highlighting its outstanding performance in predicting complex reaction pathways.
+
+2. **Osimertinib**:
+ - Osimertinib, a drug used to treat non-small-cell lung cancer, features a five-step retrosynthesis pathway.
+ - NAG2G accurately predicted each reaction type, with most predictions ranking first, showcasing its efficiency in drug molecule retrosynthesis.
+
+3. **Salmeterol**:
+ - The synthesis pathway for Salmeterol consists of five steps.
+ - NAG2G accurately predicted key steps such as protection reactions, etherification, and asymmetric Henry reactions, demonstrating its flexibility and accuracy when handling molecules with complex functional groups.
+
+4. **Lenalidomide**:
+ - Lenalidomide is a complex drug molecule with a four-step synthesis pathway.
+ - NAG2G successfully predicted reduction reactions and NBS (N-Bromosuccinimide) substitution reactions and proposed a reasonable cyclization mechanism, further showcasing its capability to predict complex reactions.
+
+*Figure 6* underscores NAG2G's advantages in high-precision prediction and multi-step reaction handling by demonstrating its application in the synthesis pathways of real-world drug molecules. These examples validate the model's reliability and accuracy while highlighting its tremendous potential in drug discovery and chemical synthesis design.
+
+
+
+*Table 4* evaluates the Top-k validity of molecules generated by NAG2G on the USPTO-50k dataset. The results demonstrate NAG2G's exceptional performance in generating valid molecules, further verifying its reliability in molecular generation tasks.
+
+---
+
+### **3.2 Practical Application Cases: Exceptional Performance of NAG2G in Drug Molecule Retrosynthesis Prediction**
+
+*Figure 6* showcases the retrosynthesis pathways for four drug molecules (**Nirmatrelvir**, **Osimertinib**, **Salmeterol**, and **Lenalidomide**) and provides detailed examples of NAG2G’s performance in predicting these pathways. These cases validate the accuracy and efficacy of NAG2G in real-world applications.
+
+1. **Nirmatrelvir**:
+ - The figure displays a six-step synthesis pathway for Nirmatrelvir.
+ - NAG2G successfully predicted all reaction steps, with the top-ranked predictions for the first three steps, highlighting its outstanding performance in predicting complex reaction pathways.
+
+2. **Osimertinib**:
+ - Osimertinib, a drug used to treat non-small-cell lung cancer, features a five-step retrosynthesis pathway.
+ - NAG2G accurately predicted each reaction type, with most predictions ranking first, showcasing its efficiency in drug molecule retrosynthesis.
+
+3. **Salmeterol**:
+ - The synthesis pathway for Salmeterol consists of five steps.
+ - NAG2G accurately predicted key steps such as protection reactions, etherification, and asymmetric Henry reactions, demonstrating its flexibility and accuracy when handling molecules with complex functional groups.
+
+4. **Lenalidomide**:
+ - Lenalidomide is a complex drug molecule with a four-step synthesis pathway.
+ - NAG2G successfully predicted reduction reactions and NBS (N-Bromosuccinimide) substitution reactions and proposed a reasonable cyclization mechanism, further showcasing its capability to predict complex reactions.
+
+*Figure 6* underscores NAG2G's advantages in high-precision prediction and multi-step reaction handling by demonstrating its application in the synthesis pathways of real-world drug molecules. These examples validate the model's reliability and accuracy while highlighting its tremendous potential in drug discovery and chemical synthesis design.
+
+ +
+## Conclusions
+This study introduces the **NAG2G model**, a template-free deep learning approach based on node alignment and a Transformer architecture for single-step retrosynthesis prediction. By integrating 2D molecular graphs and 3D conformation information, NAG2G preserves detailed molecular structure features and achieves precise product-to-reactant atom mapping through its node alignment strategy.
+
+The **Uni-Mol encoder** plays a critical role in the NAG2G model. By converting 2D and 3D information into efficient numerical representations, Uni-Mol enables the model to better understand and process molecular structures, significantly improving prediction accuracy.
+
+Experimental results demonstrate that NAG2G outperforms existing template-based and semi-template methods on both the **USPTO-50k** and **USPTO-Full** datasets, showcasing exceptional predictive performance.
+
+Ablation studies further validate the importance of node alignment, data augmentation, and time-varying graph features in enhancing model performance. By leveraging the Uni-Mol encoder, NAG2G efficiently handles and accurately predicts complex chemical reactions.
+
+Real-world case studies highlight NAG2G’s powerful capabilities in predicting synthesis pathways for drug molecules, confirming its vast potential and practical applicability in chemical synthesis design.
diff --git a/source/_posts/unimol_plus.md b/source/_posts/unimol_plus.md
new file mode 100644
index 00000000..9595d957
--- /dev/null
+++ b/source/_posts/unimol_plus.md
@@ -0,0 +1,199 @@
+---
+title: "What Can Uni-Mol Do too? | Breaking the Limits of Few-shot Molecular Property Prediction"
+date: 2024-08-21
+categories:
+- Uni-Mol
+mathjax: true
+---
+
+On August 19, 2024, Shuqi Lu and Zhifeng Gao from DP Technology, in collaboration with Professor Di He from Peking University, published a research article titled "Data-driven quantum chemical property prediction leveraging 3D conformations with Uni-Mol+" in Nature Communications. This study introduces Uni-Mol+, a deep learning algorithm that innovatively utilizes neural networks to iteratively optimize initial 3D molecular conformations, enabling precise prediction of quantum chemical properties. By progressively approximating Density Functional Theory (DFT) equilibrium conformations, Uni-Mol+ significantly enhances prediction accuracy, providing a powerful tool for high-throughput screening and new material design.
+
+
+
+## 1.Research Background
+
+Uni-Mol+ is an innovative deep learning approach designed to accelerate the prediction of quantum chemical properties. Traditional methods typically rely on 1D or 2D data for direct prediction, but these approaches often lack precision since most quantum chemical properties depend on 3D equilibrium conformations optimized via electronic structure methods such as Density Functional Theory (DFT). Uni-Mol+ employs a dual-track Transformer model architecture along with a novel training strategy to optimize conformation generation and quantum chemical property prediction.
+
+*Figure 1* illustrates the Uni-Mol+ framework:
+
+1. **Initial 3D Conformation Generation**:
+ Uni-Mol+ begins by generating initial 3D molecular conformations from 1D/2D data using low-cost tools such as RDKit or OpenBabel.
+
+2. **Iterative Refinement**:
+ The initial conformations, though generated quickly, are often not accurate enough. Uni-Mol+ iteratively refines these conformations using neural networks, progressively approximating the DFT-optimized equilibrium conformations.
+
+3. **Final Prediction**:
+ Once the conformation optimization is complete, Uni-Mol+ predicts quantum chemical properties based on the refined 3D conformations, significantly improving prediction accuracy.
+
+Experimental results demonstrate that Uni-Mol+ outperforms all existing state-of-the-art methods on several public benchmark datasets, including PCQM4MV2 and Open Catalyst 2020. This highlights its immense potential for accelerating high-throughput screening, as well as the design of new materials and molecules.
+
+
+
+## Conclusions
+This study introduces the **NAG2G model**, a template-free deep learning approach based on node alignment and a Transformer architecture for single-step retrosynthesis prediction. By integrating 2D molecular graphs and 3D conformation information, NAG2G preserves detailed molecular structure features and achieves precise product-to-reactant atom mapping through its node alignment strategy.
+
+The **Uni-Mol encoder** plays a critical role in the NAG2G model. By converting 2D and 3D information into efficient numerical representations, Uni-Mol enables the model to better understand and process molecular structures, significantly improving prediction accuracy.
+
+Experimental results demonstrate that NAG2G outperforms existing template-based and semi-template methods on both the **USPTO-50k** and **USPTO-Full** datasets, showcasing exceptional predictive performance.
+
+Ablation studies further validate the importance of node alignment, data augmentation, and time-varying graph features in enhancing model performance. By leveraging the Uni-Mol encoder, NAG2G efficiently handles and accurately predicts complex chemical reactions.
+
+Real-world case studies highlight NAG2G’s powerful capabilities in predicting synthesis pathways for drug molecules, confirming its vast potential and practical applicability in chemical synthesis design.
diff --git a/source/_posts/unimol_plus.md b/source/_posts/unimol_plus.md
new file mode 100644
index 00000000..9595d957
--- /dev/null
+++ b/source/_posts/unimol_plus.md
@@ -0,0 +1,199 @@
+---
+title: "What Can Uni-Mol Do too? | Breaking the Limits of Few-shot Molecular Property Prediction"
+date: 2024-08-21
+categories:
+- Uni-Mol
+mathjax: true
+---
+
+On August 19, 2024, Shuqi Lu and Zhifeng Gao from DP Technology, in collaboration with Professor Di He from Peking University, published a research article titled "Data-driven quantum chemical property prediction leveraging 3D conformations with Uni-Mol+" in Nature Communications. This study introduces Uni-Mol+, a deep learning algorithm that innovatively utilizes neural networks to iteratively optimize initial 3D molecular conformations, enabling precise prediction of quantum chemical properties. By progressively approximating Density Functional Theory (DFT) equilibrium conformations, Uni-Mol+ significantly enhances prediction accuracy, providing a powerful tool for high-throughput screening and new material design.
+
+
+
+## 1.Research Background
+
+Uni-Mol+ is an innovative deep learning approach designed to accelerate the prediction of quantum chemical properties. Traditional methods typically rely on 1D or 2D data for direct prediction, but these approaches often lack precision since most quantum chemical properties depend on 3D equilibrium conformations optimized via electronic structure methods such as Density Functional Theory (DFT). Uni-Mol+ employs a dual-track Transformer model architecture along with a novel training strategy to optimize conformation generation and quantum chemical property prediction.
+
+*Figure 1* illustrates the Uni-Mol+ framework:
+
+1. **Initial 3D Conformation Generation**:
+ Uni-Mol+ begins by generating initial 3D molecular conformations from 1D/2D data using low-cost tools such as RDKit or OpenBabel.
+
+2. **Iterative Refinement**:
+ The initial conformations, though generated quickly, are often not accurate enough. Uni-Mol+ iteratively refines these conformations using neural networks, progressively approximating the DFT-optimized equilibrium conformations.
+
+3. **Final Prediction**:
+ Once the conformation optimization is complete, Uni-Mol+ predicts quantum chemical properties based on the refined 3D conformations, significantly improving prediction accuracy.
+
+Experimental results demonstrate that Uni-Mol+ outperforms all existing state-of-the-art methods on several public benchmark datasets, including PCQM4MV2 and Open Catalyst 2020. This highlights its immense potential for accelerating high-throughput screening, as well as the design of new materials and molecules.
+
+ +
+## 2.Related Advances in the Field
+
+**1. Deep Learning Models Based on 1D/2D Information**
+Early studies primarily relied on 1D information (e.g., SMILES sequences) or 2D molecular graphs to predict quantum chemical properties. While these approaches are data-accessible, their predictive accuracy is often limited, as most quantum chemical properties depend on 3D equilibrium conformations optimized using Density Functional Theory (DFT).
+
+**2. Deep Learning Models Incorporating 3D Information**
+In recent years, there has been a growing trend toward incorporating 3D conformation information to enhance the performance of prediction models. For example, some methods integrate 3D structural data during training to improve the accuracy of 2D representations. However, during inference, these models still primarily rely on 2D information.
+
+**3. 3D Conformation Optimization and Prediction**
+Recent studies have focused on predicting properties directly from 3D conformations, emphasizing models' rotational and translational invariance. For instance, Uni-Mol uses 3D conformations generated by RDKit as input and introduces 3D positional recovery tasks during pretraining. However, these methods rely solely on the initial 3D conformations during inference and fail to fully optimize them to DFT-equilibrium conformations.
+
+**4. Challenges in Molecular Conformation Optimization**
+Molecular conformation optimization is a critical challenge in computational chemistry. While traditional DFT methods are accurate, they are computationally expensive. To improve efficiency, several deep learning-based potential energy models have been developed. These models use neural networks to replace DFT’s high-cost calculations but still require multiple iterations to optimize conformations.
+
+In contrast, **Uni-Mol+** achieves comparable results with only a few optimization iterations. It offers an end-to-end framework that simultaneously optimizes conformations and predicts quantum chemical properties, significantly improving efficiency while maintaining accuracy.
+
+## 3.Methods
+For any molecule, Uni-Mol+ begins by generating an initial 3D conformation using low-cost methods, such as template-based approaches from RDKit or OpenBabel. Next, it iteratively refines the initial conformation toward the target conformation, which is the DFT-optimized equilibrium conformation. Finally, based on the refined conformation, Uni-Mol+ predicts quantum chemical properties.
+
+To achieve this, a novel model architecture and a new training strategy were introduced to optimize conformations and predict quantum chemical properties. These aspects are discussed in detail in the following sections.
+
+### 3.1 Model Architecture
+
+The research team designed a novel model architecture capable of simultaneously learning molecular conformations and predicting quantum chemical properties, represented as \( (y, \hat{r}) = f(X, E, r; \theta) \). This model uses atomic features (\(X \in \mathbb{R}^{n \times d_f}\), where \(n\) is the number of atoms and \(d_f\) is the dimension of atomic features), edge features (\(E \in \mathbb{R}^{n \times n \times d_e}\), where \(d_e\) is the dimension of edge features), and atomic 3D coordinates (\(r \in \mathbb{R}^{n \times 3}\)). By optimizing the parameters \(\theta\), it predicts quantum chemical properties \(y\) and updates the 3D coordinates \(\hat{r} \in \mathbb{R}^{n \times 3}\).
+
+This architecture preserves two representation pathways similar to Uni-Mol:
+1. **Atomic representation** (\(x \in \mathbb{R}^{n \times d_x}\), where \(d_x\) is the atomic feature dimension).
+2. **Pairwise representation** (\(p \in \mathbb{R}^{n \times n \times d_p}\), where \(d_p\) is the pairwise feature dimension).
+
+The model consists of a single block, with \(x^{(l)}\) and \(p^{(l)}\) as the outputs of the \(l\)-th block.
+
+Figure 2 illustrates the overall architecture. Compared to Uni-Mol, there are several key differences:
+
+1. **Position Encoding**:
+ The model uses positional encoding methods to embed 3D spatial and 2D graph positional information. Specifically, for 3D spatial information, Gaussian kernels are used for encoding, similar to previous research. Additionally, graph positional encodings, such as shortest-path and hop encodings, are introduced.
+
+2. **Atomic Representation Update**:
+ In each block, atomic representations are iteratively updated through an attention mechanism and feed-forward neural networks.
+
+3. **Pairwise Representation Update**:
+ Pairwise representations are updated through outer products, triangle updates, and feed-forward neural networks, further enhancing the expressiveness of pairwise representations.
+
+
+
+## 2.Related Advances in the Field
+
+**1. Deep Learning Models Based on 1D/2D Information**
+Early studies primarily relied on 1D information (e.g., SMILES sequences) or 2D molecular graphs to predict quantum chemical properties. While these approaches are data-accessible, their predictive accuracy is often limited, as most quantum chemical properties depend on 3D equilibrium conformations optimized using Density Functional Theory (DFT).
+
+**2. Deep Learning Models Incorporating 3D Information**
+In recent years, there has been a growing trend toward incorporating 3D conformation information to enhance the performance of prediction models. For example, some methods integrate 3D structural data during training to improve the accuracy of 2D representations. However, during inference, these models still primarily rely on 2D information.
+
+**3. 3D Conformation Optimization and Prediction**
+Recent studies have focused on predicting properties directly from 3D conformations, emphasizing models' rotational and translational invariance. For instance, Uni-Mol uses 3D conformations generated by RDKit as input and introduces 3D positional recovery tasks during pretraining. However, these methods rely solely on the initial 3D conformations during inference and fail to fully optimize them to DFT-equilibrium conformations.
+
+**4. Challenges in Molecular Conformation Optimization**
+Molecular conformation optimization is a critical challenge in computational chemistry. While traditional DFT methods are accurate, they are computationally expensive. To improve efficiency, several deep learning-based potential energy models have been developed. These models use neural networks to replace DFT’s high-cost calculations but still require multiple iterations to optimize conformations.
+
+In contrast, **Uni-Mol+** achieves comparable results with only a few optimization iterations. It offers an end-to-end framework that simultaneously optimizes conformations and predicts quantum chemical properties, significantly improving efficiency while maintaining accuracy.
+
+## 3.Methods
+For any molecule, Uni-Mol+ begins by generating an initial 3D conformation using low-cost methods, such as template-based approaches from RDKit or OpenBabel. Next, it iteratively refines the initial conformation toward the target conformation, which is the DFT-optimized equilibrium conformation. Finally, based on the refined conformation, Uni-Mol+ predicts quantum chemical properties.
+
+To achieve this, a novel model architecture and a new training strategy were introduced to optimize conformations and predict quantum chemical properties. These aspects are discussed in detail in the following sections.
+
+### 3.1 Model Architecture
+
+The research team designed a novel model architecture capable of simultaneously learning molecular conformations and predicting quantum chemical properties, represented as \( (y, \hat{r}) = f(X, E, r; \theta) \). This model uses atomic features (\(X \in \mathbb{R}^{n \times d_f}\), where \(n\) is the number of atoms and \(d_f\) is the dimension of atomic features), edge features (\(E \in \mathbb{R}^{n \times n \times d_e}\), where \(d_e\) is the dimension of edge features), and atomic 3D coordinates (\(r \in \mathbb{R}^{n \times 3}\)). By optimizing the parameters \(\theta\), it predicts quantum chemical properties \(y\) and updates the 3D coordinates \(\hat{r} \in \mathbb{R}^{n \times 3}\).
+
+This architecture preserves two representation pathways similar to Uni-Mol:
+1. **Atomic representation** (\(x \in \mathbb{R}^{n \times d_x}\), where \(d_x\) is the atomic feature dimension).
+2. **Pairwise representation** (\(p \in \mathbb{R}^{n \times n \times d_p}\), where \(d_p\) is the pairwise feature dimension).
+
+The model consists of a single block, with \(x^{(l)}\) and \(p^{(l)}\) as the outputs of the \(l\)-th block.
+
+Figure 2 illustrates the overall architecture. Compared to Uni-Mol, there are several key differences:
+
+1. **Position Encoding**:
+ The model uses positional encoding methods to embed 3D spatial and 2D graph positional information. Specifically, for 3D spatial information, Gaussian kernels are used for encoding, similar to previous research. Additionally, graph positional encodings, such as shortest-path and hop encodings, are introduced.
+
+2. **Atomic Representation Update**:
+ In each block, atomic representations are iteratively updated through an attention mechanism and feed-forward neural networks.
+
+3. **Pairwise Representation Update**:
+ Pairwise representations are updated through outer products, triangle updates, and feed-forward neural networks, further enhancing the expressiveness of pairwise representations.
+
+ +
+### 3.2 Model Training
+In DFT conformation optimization or molecular dynamics simulations, conformations are iteratively optimized, forming a trajectory from the initial conformation to the equilibrium conformation. However, saving such trajectories can be very costly, and public datasets typically provide only equilibrium conformations. To fully leverage this information, we propose a novel training method that first generates a pseudo-trajectory, samples a conformation from it as input, and uses it to predict the equilibrium conformation.
+
+## 4.**Experimental Validation of Uni-Mol+'s Exceptional Performance**
+
+In this study, the research team conducted a thorough empirical analysis of the Uni-Mol+ model’s performance. The team first detailed the model's configuration and then performed comprehensive benchmarking using two widely recognized public datasets, **PCQM4MV2** and **OC20**, to evaluate the model’s capabilities in small organic molecules and catalytic systems. Finally, ablation studies were conducted to analyze the impact of different model components and training strategies on overall performance, further confirming the superiority of Uni-Mol+.
+
+---
+
+### 4.1 **Model Configuration**
+
+The Uni-Mol+ model adopts a 12-layer architecture, with the following configurations:
+- **Atom representation**: Dimension = 768
+- **Pair representation**: Dimension = 256
+- **FFN hidden layer dimensions**:
+ - Atom representation track: 768
+ - Pair representation track: 256
+- **OuterProduct hidden dimension**: 32
+- **TriangularUpdate hidden dimension**: 32
+
+The model performs a single conformation optimization iteration, indicating a total of two iterations: one for conformation optimization and one for quantum chemical property prediction.
+
+**Training strategy**:
+- Standard deviation of random noise: 0.2
+- Sampling of parameter \( q \):
+ - \( q = 0.0 \) with probability 0.8
+ - \( q = 1.0 \) with probability 0.1
+ - Uniform sampling from [0.4, 0.6] with probability 0.1
+- Total model parameters: approximately 52.4M
+
+---
+
+### 4.2 **PCQM4MV2 Dataset**
+
+**Background**:
+The PCQM4MV2 dataset originates from the OGB Large-Scale Challenge and aims to advance and evaluate machine learning models for molecular quantum chemical property prediction, particularly for the **HOMO-LUMO gap**, which represents the energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The dataset consists of approximately 4 million molecules represented using SMILES. While the training and validation sets include HOMO-LUMO gap labels, the test set labels remain undisclosed. The training set also provides DFT-optimized equilibrium conformations, but the validation and test sets do not. The benchmarking objective is to predict the HOMO-LUMO gap using SMILES during inference, without relying on DFT equilibrium conformations.
+
+**Experimental Setup**:
+- **Initial conformations**: 8 initial conformations per molecule were generated using RDKit (approx. 0.01 seconds per molecule).
+- **Training**: During each epoch, one conformation was randomly sampled as input.
+- **Inference**: The average HOMO-LUMO gap prediction over 8 conformations was used.
+
+**Hyperparameters**:
+- Optimizer: AdamW
+- Learning rate: \( 2 \times 10^{-4} \)
+- Batch size: 1024
+- \( \beta_1 = 0.9 \), \( \beta_2 = 0.999 \)
+- Gradient clipping: 5.0
+- Training duration: 1.5 million steps (including 150k warm-up steps)
+- Exponential Moving Average (EMA) decay rate: 0.999
+
+**Training Infrastructure**:
+- **Training**: 8 NVIDIA A100 GPUs, approximately 5 days.
+- **Inference**: 8 NVIDIA V100 GPUs, approximately 7 minutes for the 147k test-dev set.
+
+**Performance Results on PCQM4MV2**
+
+The team compared the previous leaderboard submissions on PCQM4MV2 as baselines and evaluated three variants of Uni-Mol+ models (6-layer, 12-layer, and 18-layer) to study the impact of model size on performance.
+
+**Results Summary (Table 1)**:
+1. All three Uni-Mol+ variants achieved significant performance improvements over previous baseline methods.
+2. Even the 6-layer Uni-Mol+ model, despite having fewer parameters, outperformed all prior baseline methods.
+3. Increasing the number of layers from 6 to 12 resulted in a substantial accuracy improvement, far exceeding all baseline methods.
+4. The 18-layer Uni-Mol+ model delivered the best performance, achieving a significant edge over all baselines.
+
+
+
+### 3.2 Model Training
+In DFT conformation optimization or molecular dynamics simulations, conformations are iteratively optimized, forming a trajectory from the initial conformation to the equilibrium conformation. However, saving such trajectories can be very costly, and public datasets typically provide only equilibrium conformations. To fully leverage this information, we propose a novel training method that first generates a pseudo-trajectory, samples a conformation from it as input, and uses it to predict the equilibrium conformation.
+
+## 4.**Experimental Validation of Uni-Mol+'s Exceptional Performance**
+
+In this study, the research team conducted a thorough empirical analysis of the Uni-Mol+ model’s performance. The team first detailed the model's configuration and then performed comprehensive benchmarking using two widely recognized public datasets, **PCQM4MV2** and **OC20**, to evaluate the model’s capabilities in small organic molecules and catalytic systems. Finally, ablation studies were conducted to analyze the impact of different model components and training strategies on overall performance, further confirming the superiority of Uni-Mol+.
+
+---
+
+### 4.1 **Model Configuration**
+
+The Uni-Mol+ model adopts a 12-layer architecture, with the following configurations:
+- **Atom representation**: Dimension = 768
+- **Pair representation**: Dimension = 256
+- **FFN hidden layer dimensions**:
+ - Atom representation track: 768
+ - Pair representation track: 256
+- **OuterProduct hidden dimension**: 32
+- **TriangularUpdate hidden dimension**: 32
+
+The model performs a single conformation optimization iteration, indicating a total of two iterations: one for conformation optimization and one for quantum chemical property prediction.
+
+**Training strategy**:
+- Standard deviation of random noise: 0.2
+- Sampling of parameter \( q \):
+ - \( q = 0.0 \) with probability 0.8
+ - \( q = 1.0 \) with probability 0.1
+ - Uniform sampling from [0.4, 0.6] with probability 0.1
+- Total model parameters: approximately 52.4M
+
+---
+
+### 4.2 **PCQM4MV2 Dataset**
+
+**Background**:
+The PCQM4MV2 dataset originates from the OGB Large-Scale Challenge and aims to advance and evaluate machine learning models for molecular quantum chemical property prediction, particularly for the **HOMO-LUMO gap**, which represents the energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The dataset consists of approximately 4 million molecules represented using SMILES. While the training and validation sets include HOMO-LUMO gap labels, the test set labels remain undisclosed. The training set also provides DFT-optimized equilibrium conformations, but the validation and test sets do not. The benchmarking objective is to predict the HOMO-LUMO gap using SMILES during inference, without relying on DFT equilibrium conformations.
+
+**Experimental Setup**:
+- **Initial conformations**: 8 initial conformations per molecule were generated using RDKit (approx. 0.01 seconds per molecule).
+- **Training**: During each epoch, one conformation was randomly sampled as input.
+- **Inference**: The average HOMO-LUMO gap prediction over 8 conformations was used.
+
+**Hyperparameters**:
+- Optimizer: AdamW
+- Learning rate: \( 2 \times 10^{-4} \)
+- Batch size: 1024
+- \( \beta_1 = 0.9 \), \( \beta_2 = 0.999 \)
+- Gradient clipping: 5.0
+- Training duration: 1.5 million steps (including 150k warm-up steps)
+- Exponential Moving Average (EMA) decay rate: 0.999
+
+**Training Infrastructure**:
+- **Training**: 8 NVIDIA A100 GPUs, approximately 5 days.
+- **Inference**: 8 NVIDIA V100 GPUs, approximately 7 minutes for the 147k test-dev set.
+
+**Performance Results on PCQM4MV2**
+
+The team compared the previous leaderboard submissions on PCQM4MV2 as baselines and evaluated three variants of Uni-Mol+ models (6-layer, 12-layer, and 18-layer) to study the impact of model size on performance.
+
+**Results Summary (Table 1)**:
+1. All three Uni-Mol+ variants achieved significant performance improvements over previous baseline methods.
+2. Even the 6-layer Uni-Mol+ model, despite having fewer parameters, outperformed all prior baseline methods.
+3. Increasing the number of layers from 6 to 12 resulted in a substantial accuracy improvement, far exceeding all baseline methods.
+4. The 18-layer Uni-Mol+ model delivered the best performance, achieving a significant edge over all baselines.
+
+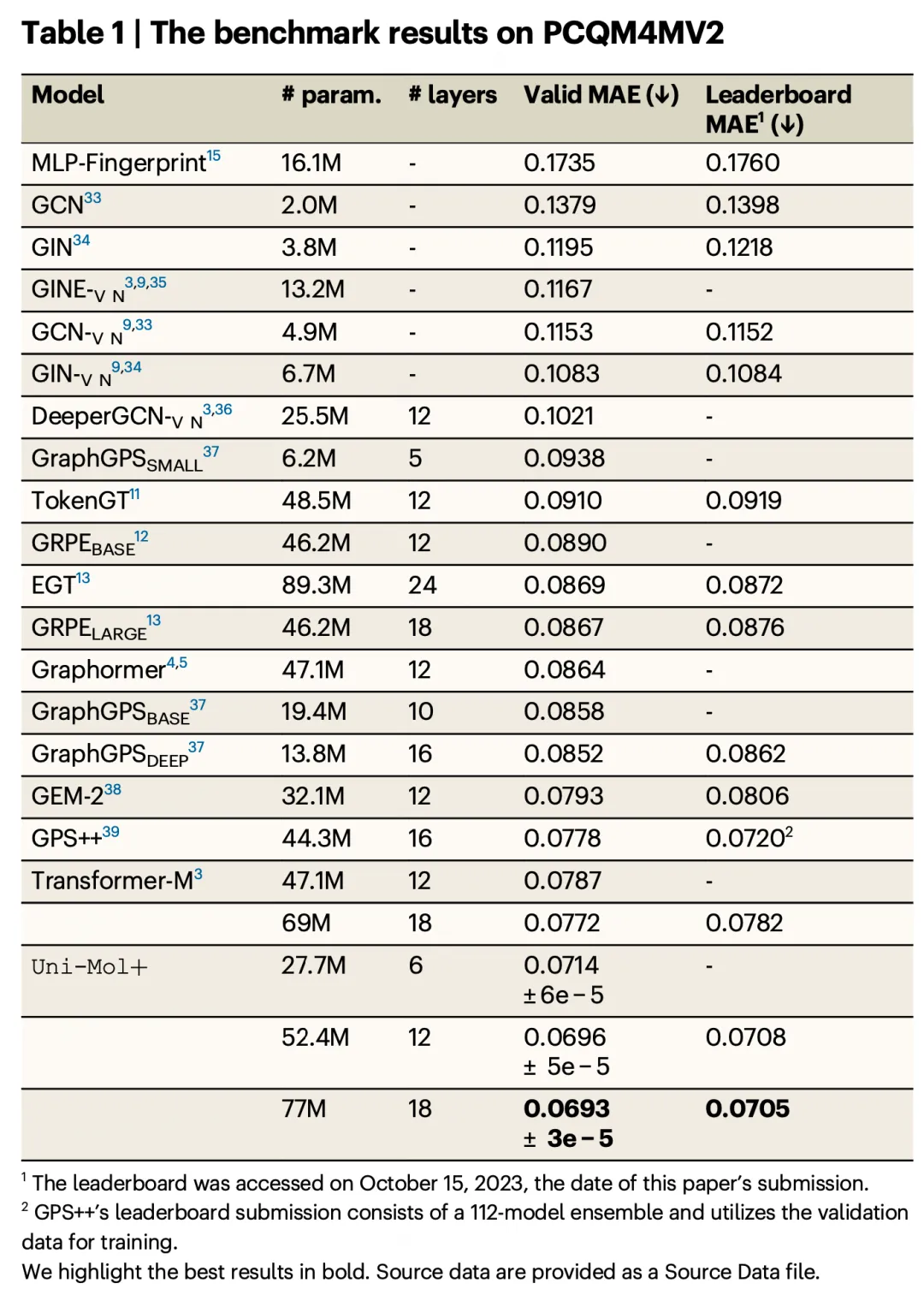 +
+
+### 4.3 **Open Catalyst 2020**
+
+The **Open Catalyst 2020 (OC20)** dataset is designed to advance machine learning models for catalyst discovery and optimization. OC20 includes three tasks: **Structure to Energy and Forces (S2EF)**, **Initial Structure to Relaxed Structure (IS2RS)**, and **Initial Structure to Relaxed Energy (IS2RE)**. This study focuses on the IS2RE task, which aligns with the objectives of the proposed method. The IS2RE task aims to predict the relaxed energy based on the initial conformation. The dataset contains approximately 460,000 training data points. While DFT-equilibrium conformations are available during training, their use is prohibited during inference. Unlike the PCQM4MV2 dataset, the initial conformations are already provided in OC20, eliminating the need to generate initial input conformations.
+
+**Experimental Setup**
+
+For the OC20 experiments, the team used the default 12-layer Uni-Mol+ configuration with some modifications compared to the PCQM4MV2 setup:
+
+1. **Exclusion of Graph Features**:
+ Since the OC20 dataset lacks graph information, all graph-related features were excluded from the model.
+
+2. **Model Capacity Adjustment**:
+ Due to the larger number of atoms in the OC20 dataset compared to PCQM4MV2, the model capacity was slightly reduced for efficiency. Specifically:
+ - **Pair representation dimension**: 128
+ - **OuterProduct hidden dimension**: 16
+ - **TriangularUpdate hidden dimension**: 16
+
+3. **Periodic Boundary Conditions**:
+ To account for periodic boundary conditions, the team employed a pre-expansion of neighboring cells combined with a radius cutoff to reduce the number of atoms considered.
+
+4. **Training Configuration**:
+ - Optimizer: AdamW
+ - Training steps: 1.5 million (including 150,000 warm-up steps)
+ - Learning rate: \(2 \times 10^{-4}\)
+ - Batch size: 64
+ - \( \beta_1 = 0.9 \), \( \beta_2 = 0.999 \)
+ - Gradient clipping: 5.0
+
+The training process took approximately 7 days using 16 NVIDIA A100 GPUs.
+
+**Results**
+
+*Tables 2 and 3* present the performance comparisons of various models on the OC20 IS2RE validation and test sets. Metrics include:
+- **Mean Absolute Error (MAE)** for energy predictions
+- **Energy within Threshold (EwT%)**, which measures the percentage of energy predictions falling within a certain threshold.
+
+**Key Findings**:
+1. **Significant Improvements**: Uni-Mol+ outperformed all prior baseline methods in both MAE and EwT across all categories.
+2. **Lowest MAE Across Categories**:
+ - Uni-Mol+ achieved the lowest MAE values across all categories, including:
+ - In-domain (ID)
+ - Out-of-domain Adsorption (OOD Ads.)
+ - Out-of-domain Catalyst (OOD Cat.)
+ - Out-of-domain Both (OOD Both)
+ - Average (AVG)
+3. **Highest EwT Scores**:
+ - Uni-Mol+ also achieved the highest EwT scores in all categories, emphasizing its robustness across both in-domain and out-of-domain data.
+
+
+
+
+### 4.3 **Open Catalyst 2020**
+
+The **Open Catalyst 2020 (OC20)** dataset is designed to advance machine learning models for catalyst discovery and optimization. OC20 includes three tasks: **Structure to Energy and Forces (S2EF)**, **Initial Structure to Relaxed Structure (IS2RS)**, and **Initial Structure to Relaxed Energy (IS2RE)**. This study focuses on the IS2RE task, which aligns with the objectives of the proposed method. The IS2RE task aims to predict the relaxed energy based on the initial conformation. The dataset contains approximately 460,000 training data points. While DFT-equilibrium conformations are available during training, their use is prohibited during inference. Unlike the PCQM4MV2 dataset, the initial conformations are already provided in OC20, eliminating the need to generate initial input conformations.
+
+**Experimental Setup**
+
+For the OC20 experiments, the team used the default 12-layer Uni-Mol+ configuration with some modifications compared to the PCQM4MV2 setup:
+
+1. **Exclusion of Graph Features**:
+ Since the OC20 dataset lacks graph information, all graph-related features were excluded from the model.
+
+2. **Model Capacity Adjustment**:
+ Due to the larger number of atoms in the OC20 dataset compared to PCQM4MV2, the model capacity was slightly reduced for efficiency. Specifically:
+ - **Pair representation dimension**: 128
+ - **OuterProduct hidden dimension**: 16
+ - **TriangularUpdate hidden dimension**: 16
+
+3. **Periodic Boundary Conditions**:
+ To account for periodic boundary conditions, the team employed a pre-expansion of neighboring cells combined with a radius cutoff to reduce the number of atoms considered.
+
+4. **Training Configuration**:
+ - Optimizer: AdamW
+ - Training steps: 1.5 million (including 150,000 warm-up steps)
+ - Learning rate: \(2 \times 10^{-4}\)
+ - Batch size: 64
+ - \( \beta_1 = 0.9 \), \( \beta_2 = 0.999 \)
+ - Gradient clipping: 5.0
+
+The training process took approximately 7 days using 16 NVIDIA A100 GPUs.
+
+**Results**
+
+*Tables 2 and 3* present the performance comparisons of various models on the OC20 IS2RE validation and test sets. Metrics include:
+- **Mean Absolute Error (MAE)** for energy predictions
+- **Energy within Threshold (EwT%)**, which measures the percentage of energy predictions falling within a certain threshold.
+
+**Key Findings**:
+1. **Significant Improvements**: Uni-Mol+ outperformed all prior baseline methods in both MAE and EwT across all categories.
+2. **Lowest MAE Across Categories**:
+ - Uni-Mol+ achieved the lowest MAE values across all categories, including:
+ - In-domain (ID)
+ - Out-of-domain Adsorption (OOD Ads.)
+ - Out-of-domain Catalyst (OOD Cat.)
+ - Out-of-domain Both (OOD Both)
+ - Average (AVG)
+3. **Highest EwT Scores**:
+ - Uni-Mol+ also achieved the highest EwT scores in all categories, emphasizing its robustness across both in-domain and out-of-domain data.
+
+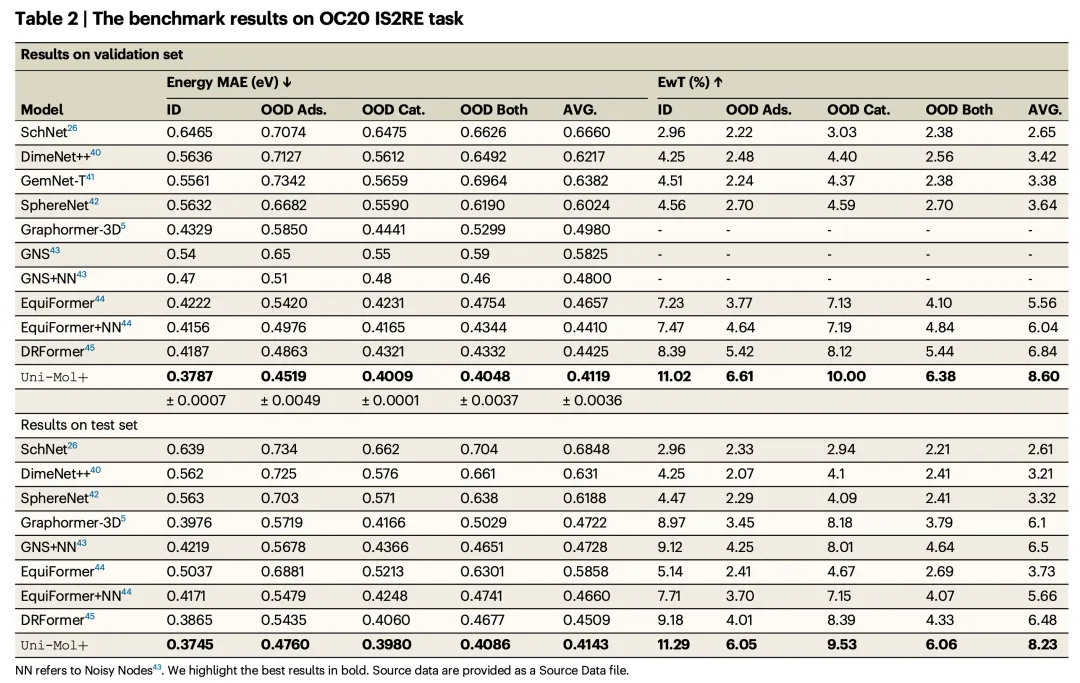 +
+## Conclusions
+In this study, the Uni-Mol+ model introduces a novel approach to quantum chemical property prediction using deep learning. Unlike traditional methods, Uni-Mol+ begins by leveraging low-cost tools to generate initial 3D conformations from 1D/2D data. These conformations are then iteratively optimized to DFT-equilibrium conformations through neural networks, and quantum chemical properties are predicted based on the optimized conformations.
+
+The results demonstrate that Uni-Mol+ significantly outperforms all previous methods across multiple public benchmark datasets, including PCQM4MV2 and Open Catalyst 2020. Ablation studies further validate the effectiveness of each component of the model.
+
+Overall, Uni-Mol+ showcases immense potential for accelerating quantum chemical property prediction through deep learning, providing an effective tool for high-throughput screening, new material discovery, and novel molecule design.
\ No newline at end of file
diff --git a/source/_posts/unimol_small.md b/source/_posts/unimol_small.md
new file mode 100644
index 00000000..8b80b756
--- /dev/null
+++ b/source/_posts/unimol_small.md
@@ -0,0 +1,127 @@
+---
+title: "What Can Uni-Mol Do too? | Breaking the Limits of Few-shot Molecular Property Prediction"
+date: 2024-12-02
+categories:
+- Uni-Mol
+mathjax: true
+---
+
+On August 19, 2024, Linlin Hou, Hongxin Xiang, and Xiangxiang Zeng from Hunan University, in collaboration with Li Zeng from the Shanghai Institute of Materia Medica, published a research article titled *"Attribute-guided Prototype Network for Few-shot Molecular Property Prediction"* in *Briefings in Bioinformatics*. This study introduced an Attribute-guided Prototype Network (APN), which innovatively combines high-level molecular fingerprints with deep learning algorithms, significantly improving the accuracy of molecular property prediction in limited-sample scenarios. This breakthrough opens a new direction for few-shot learning in drug development.
+
+
+
+## Research Background
+Molecular property prediction (MPP) is a critical step in the drug discovery process. However, traditional deep learning methods often rely on large amounts of labeled data, making their application in few-shot scenarios highly challenging. Recently, a research team proposed an innovative Attribute-guided Prototype Network (APN), which overcomes the limitations of few-shot learning by extracting and leveraging high-dimensional molecular fingerprint attributes. Compared to existing methods, APN not only efficiently captures both local and global information within molecular structures but also demonstrates stronger generalization capabilities. This study offers new possibilities for molecular property prediction under limited data conditions, significantly accelerating virtual screening and drug optimization processes.
+
+## Uni-Mol Assists in Few-shot Molecular Property Prediction
+
+In this study, Uni-Mol, a self-supervised learning model, was employed to extract deep attributes from molecular structures. These deep attributes, utilized in training neural networks, enabled the model to capture high-level molecular features, providing strong support for few-shot molecular property prediction. Specifically, Uni-Mol generated multiple molecular conformations (using 10 conformations in unimol_10conf) and, combined with Graph Attention Networks (GAT) and other molecular fingerprint data, optimized the local and global representations of molecules.
+
+Experimental results demonstrated that Uni-Mol achieved outstanding performance on several datasets, including Tox21, significantly enhancing the prediction accuracy in few-shot learning scenarios.
+
+## Methods
+**Attribute-guided Prototype Network (APN)**
+
+
+
+
+## Conclusions
+In this study, the Uni-Mol+ model introduces a novel approach to quantum chemical property prediction using deep learning. Unlike traditional methods, Uni-Mol+ begins by leveraging low-cost tools to generate initial 3D conformations from 1D/2D data. These conformations are then iteratively optimized to DFT-equilibrium conformations through neural networks, and quantum chemical properties are predicted based on the optimized conformations.
+
+The results demonstrate that Uni-Mol+ significantly outperforms all previous methods across multiple public benchmark datasets, including PCQM4MV2 and Open Catalyst 2020. Ablation studies further validate the effectiveness of each component of the model.
+
+Overall, Uni-Mol+ showcases immense potential for accelerating quantum chemical property prediction through deep learning, providing an effective tool for high-throughput screening, new material discovery, and novel molecule design.
\ No newline at end of file
diff --git a/source/_posts/unimol_small.md b/source/_posts/unimol_small.md
new file mode 100644
index 00000000..8b80b756
--- /dev/null
+++ b/source/_posts/unimol_small.md
@@ -0,0 +1,127 @@
+---
+title: "What Can Uni-Mol Do too? | Breaking the Limits of Few-shot Molecular Property Prediction"
+date: 2024-12-02
+categories:
+- Uni-Mol
+mathjax: true
+---
+
+On August 19, 2024, Linlin Hou, Hongxin Xiang, and Xiangxiang Zeng from Hunan University, in collaboration with Li Zeng from the Shanghai Institute of Materia Medica, published a research article titled *"Attribute-guided Prototype Network for Few-shot Molecular Property Prediction"* in *Briefings in Bioinformatics*. This study introduced an Attribute-guided Prototype Network (APN), which innovatively combines high-level molecular fingerprints with deep learning algorithms, significantly improving the accuracy of molecular property prediction in limited-sample scenarios. This breakthrough opens a new direction for few-shot learning in drug development.
+
+
+
+## Research Background
+Molecular property prediction (MPP) is a critical step in the drug discovery process. However, traditional deep learning methods often rely on large amounts of labeled data, making their application in few-shot scenarios highly challenging. Recently, a research team proposed an innovative Attribute-guided Prototype Network (APN), which overcomes the limitations of few-shot learning by extracting and leveraging high-dimensional molecular fingerprint attributes. Compared to existing methods, APN not only efficiently captures both local and global information within molecular structures but also demonstrates stronger generalization capabilities. This study offers new possibilities for molecular property prediction under limited data conditions, significantly accelerating virtual screening and drug optimization processes.
+
+## Uni-Mol Assists in Few-shot Molecular Property Prediction
+
+In this study, Uni-Mol, a self-supervised learning model, was employed to extract deep attributes from molecular structures. These deep attributes, utilized in training neural networks, enabled the model to capture high-level molecular features, providing strong support for few-shot molecular property prediction. Specifically, Uni-Mol generated multiple molecular conformations (using 10 conformations in unimol_10conf) and, combined with Graph Attention Networks (GAT) and other molecular fingerprint data, optimized the local and global representations of molecules.
+
+Experimental results demonstrated that Uni-Mol achieved outstanding performance on several datasets, including Tox21, significantly enhancing the prediction accuracy in few-shot learning scenarios.
+
+## Methods
+**Attribute-guided Prototype Network (APN)**
+
+
+ +
+**Attribute-guided Prototype Network (APN) Architecture**
+
+
+
+**Attribute-guided Prototype Network (APN) Architecture**
+
+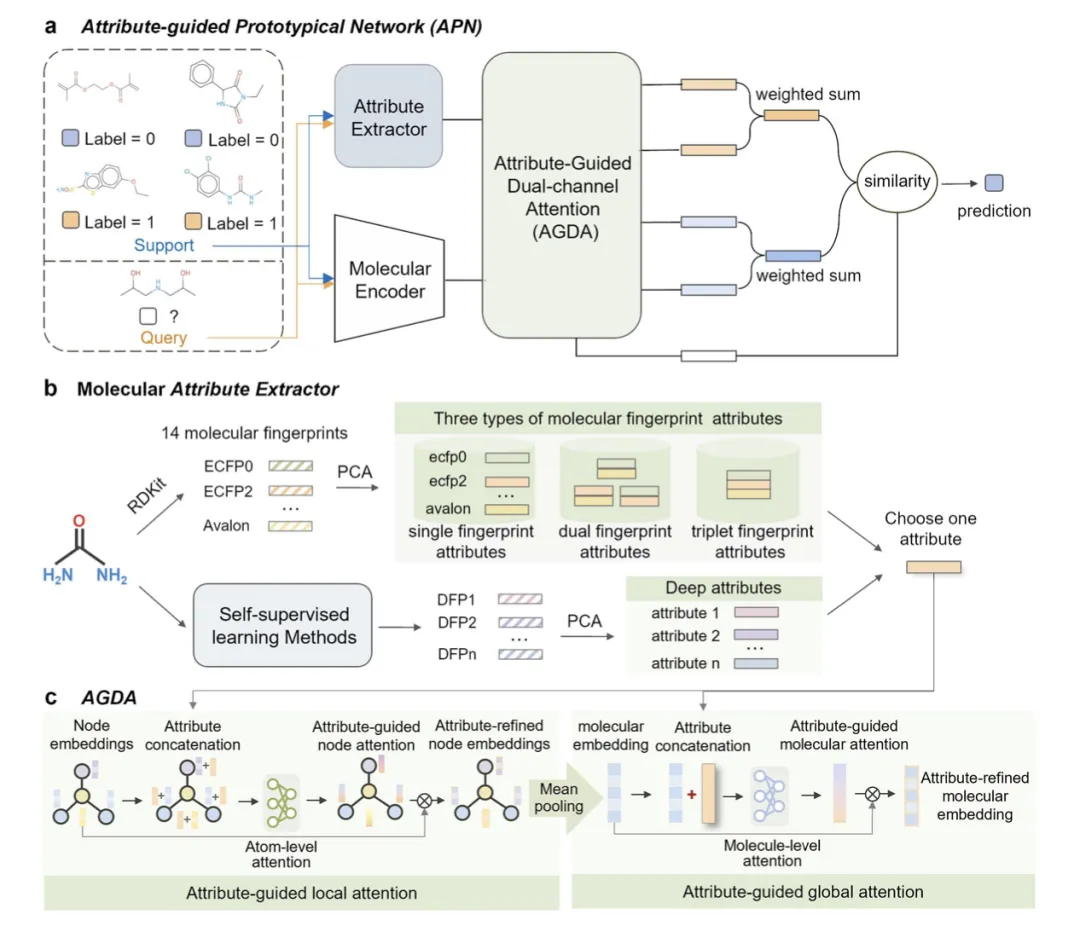 +
+
+*Figure 2* introduces the overall architecture of the Attribute-guided Prototype Network (APN), consisting of a molecular attribute extractor and an Attribute-Guided Dual-channel Attention (AGDA) module.
+
+1. **Figure 2(a) - Overall APN Architecture**:
+ This panel illustrates the APN architecture using a 2-class, 2-shot task from the Tox21 dataset as an example. APN is optimized over a series of training tasks. In each task, a support set is used to derive prototypes for each class, while a query set is used to optimize the parameters of the molecular encoder and the AGDA module. The query molecule, represented as \( z' \), is generated via the molecular encoder and AGDA module, and its similarity to the prototypes is computed for final prediction.
+
+2. **Figure 2(b) - Molecular Attribute Extractor**:
+ This panel shows how single-fingerprint, double-fingerprint, and triple-fingerprint attributes are extracted from 14 types of molecular fingerprints. These attributes are further enriched using self-supervised learning methods to derive deep attributes, which are used to optimize molecular representations.
+
+3. **Figure 2(c) - Detailed Structure of the AGDA Module**:
+ The AGDA module employs both local and global attention mechanisms to optimize atomic-level and molecular-level representations.
+ - The **local attention module** guides the model to focus on important local information.
+ - The **global attention module** helps the model capture overall molecular details.
+ Together, these mechanisms refine the molecular representation, improving its discriminative power for classification tasks.
+
+This architecture effectively combines local and global molecular attributes to enhance performance in few-shot molecular property prediction.
+
+## Results and Discussion
+**Experimental Validation and Performance Comparison of APN with Baseline Models**
+
+
+
+
+*Figure 2* introduces the overall architecture of the Attribute-guided Prototype Network (APN), consisting of a molecular attribute extractor and an Attribute-Guided Dual-channel Attention (AGDA) module.
+
+1. **Figure 2(a) - Overall APN Architecture**:
+ This panel illustrates the APN architecture using a 2-class, 2-shot task from the Tox21 dataset as an example. APN is optimized over a series of training tasks. In each task, a support set is used to derive prototypes for each class, while a query set is used to optimize the parameters of the molecular encoder and the AGDA module. The query molecule, represented as \( z' \), is generated via the molecular encoder and AGDA module, and its similarity to the prototypes is computed for final prediction.
+
+2. **Figure 2(b) - Molecular Attribute Extractor**:
+ This panel shows how single-fingerprint, double-fingerprint, and triple-fingerprint attributes are extracted from 14 types of molecular fingerprints. These attributes are further enriched using self-supervised learning methods to derive deep attributes, which are used to optimize molecular representations.
+
+3. **Figure 2(c) - Detailed Structure of the AGDA Module**:
+ The AGDA module employs both local and global attention mechanisms to optimize atomic-level and molecular-level representations.
+ - The **local attention module** guides the model to focus on important local information.
+ - The **global attention module** helps the model capture overall molecular details.
+ Together, these mechanisms refine the molecular representation, improving its discriminative power for classification tasks.
+
+This architecture effectively combines local and global molecular attributes to enhance performance in few-shot molecular property prediction.
+
+## Results and Discussion
+**Experimental Validation and Performance Comparison of APN with Baseline Models**
+
+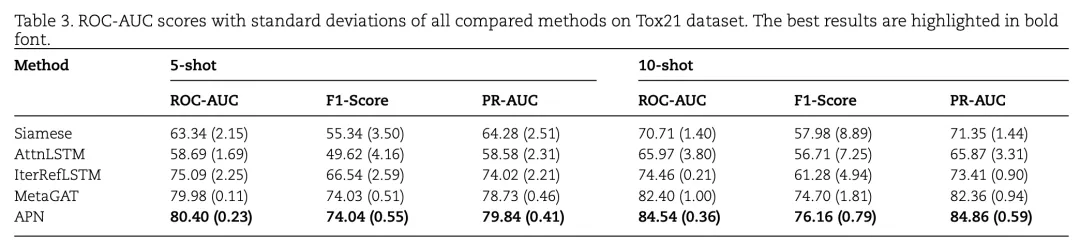 +
+
+*Table 3* compares the performance of the APN model on the Tox21 dataset with several baseline models, including Siamese networks, Attention LSTM, Iterative LSTM, and MetaGAT, for 5-shot and 10-shot tasks. The results show:
+
+- **5-shot Task**: APN achieves a ROC-AUC score of 80.40%, outperforming all other models.
+- **10-shot Task**: APN reaches a ROC-AUC score of 84.54%, again surpassing all baseline models.
+- APN also leads in F1-Score and PR-AUC scores across multiple tasks, particularly excelling in the 10-shot task.
+
+---
+
+**Analysis of Single-Fingerprint Attributes in APN**
+
+
+
+*Table 3* compares the performance of the APN model on the Tox21 dataset with several baseline models, including Siamese networks, Attention LSTM, Iterative LSTM, and MetaGAT, for 5-shot and 10-shot tasks. The results show:
+
+- **5-shot Task**: APN achieves a ROC-AUC score of 80.40%, outperforming all other models.
+- **10-shot Task**: APN reaches a ROC-AUC score of 84.54%, again surpassing all baseline models.
+- APN also leads in F1-Score and PR-AUC scores across multiple tasks, particularly excelling in the 10-shot task.
+
+---
+
+**Analysis of Single-Fingerprint Attributes in APN**
+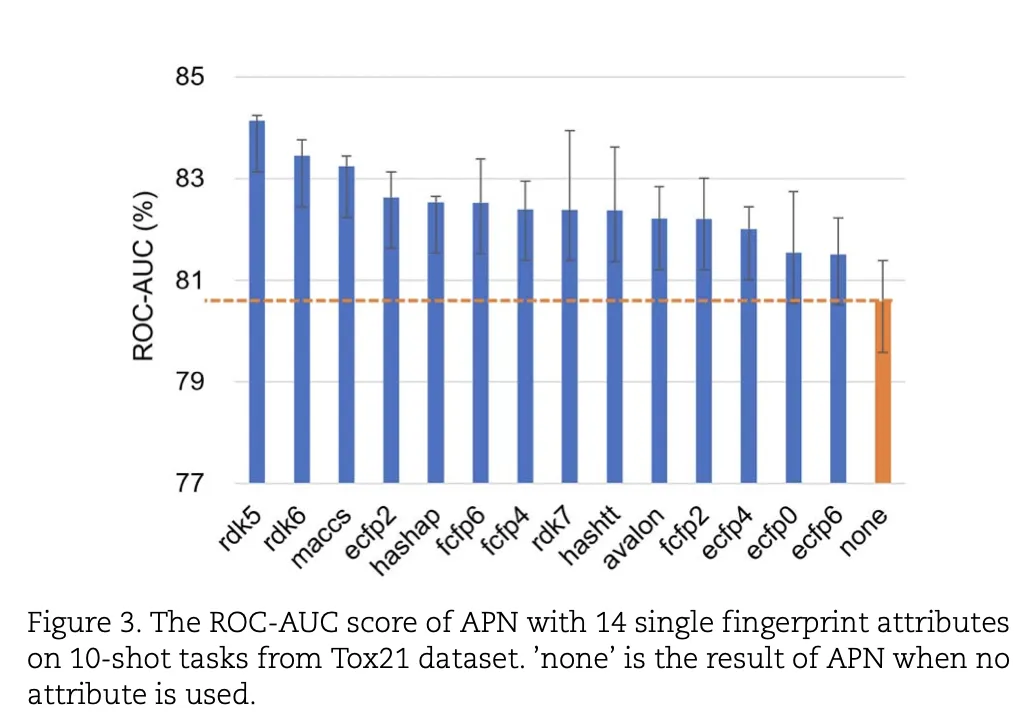 +
+*Figure 3* presents the ROC-AUC scores of APN when using 14 different single-fingerprint attributes for the Tox21 dataset in the 10-shot task:
+
+- Each bar represents the ROC-AUC score for a single fingerprint attribute.
+- Compared to the baseline without any attributes (“none”), using single-fingerprint attributes significantly improves APN's performance, with a maximum gain of 3.55%.
+- Fingerprint attributes such as RDK5, RDK6, and HashAP, which are path-based, perform exceptionally well, significantly enhancing prediction accuracy.
+
+---
+
+**Deep Attributes in APN**
+
+
+*Figure 3* presents the ROC-AUC scores of APN when using 14 different single-fingerprint attributes for the Tox21 dataset in the 10-shot task:
+
+- Each bar represents the ROC-AUC score for a single fingerprint attribute.
+- Compared to the baseline without any attributes (“none”), using single-fingerprint attributes significantly improves APN's performance, with a maximum gain of 3.55%.
+- Fingerprint attributes such as RDK5, RDK6, and HashAP, which are path-based, perform exceptionally well, significantly enhancing prediction accuracy.
+
+---
+
+**Deep Attributes in APN**
+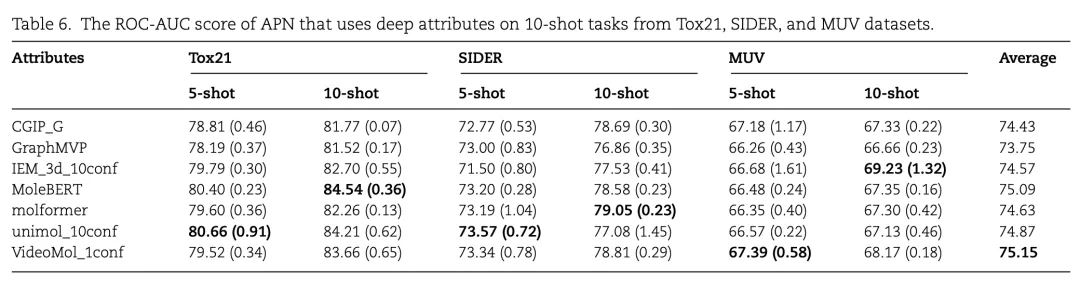 +
+*Table 6* demonstrates the performance of APN on the Tox21, SIDER, and MUV datasets using different deep attributes generated via self-supervised learning. Key findings:
+
+- **Tox21 Dataset**:
+ - Using the unimol_10conf deep attribute, APN achieves a ROC-AUC of 84.21%, F1-Score of 73.57%, and PR-AUC of 77.08%, showcasing strong predictive capability.
+
+- **SIDER Dataset**:
+ - The best-performing deep attribute is CGIP_G, with a ROC-AUC of 78.69%, while other attributes yield similar results.
+
+- **MUV Dataset**:
+ - The IEM_3d_10conf deep attribute achieves a PR-AUC of 69.23%, highlighting its potential for virtual screening tasks.
+
+These results confirm that deep attributes effectively capture complex relationships between molecular structures and properties. In some cases, they even outperform traditional molecular fingerprint attributes, demonstrating APN’s strong generalization capability in few-shot molecular property prediction tasks.
+
+---
+
+**Ablation Study on APN Modules**
+
+
+*Table 6* demonstrates the performance of APN on the Tox21, SIDER, and MUV datasets using different deep attributes generated via self-supervised learning. Key findings:
+
+- **Tox21 Dataset**:
+ - Using the unimol_10conf deep attribute, APN achieves a ROC-AUC of 84.21%, F1-Score of 73.57%, and PR-AUC of 77.08%, showcasing strong predictive capability.
+
+- **SIDER Dataset**:
+ - The best-performing deep attribute is CGIP_G, with a ROC-AUC of 78.69%, while other attributes yield similar results.
+
+- **MUV Dataset**:
+ - The IEM_3d_10conf deep attribute achieves a PR-AUC of 69.23%, highlighting its potential for virtual screening tasks.
+
+These results confirm that deep attributes effectively capture complex relationships between molecular structures and properties. In some cases, they even outperform traditional molecular fingerprint attributes, demonstrating APN’s strong generalization capability in few-shot molecular property prediction tasks.
+
+---
+
+**Ablation Study on APN Modules**
+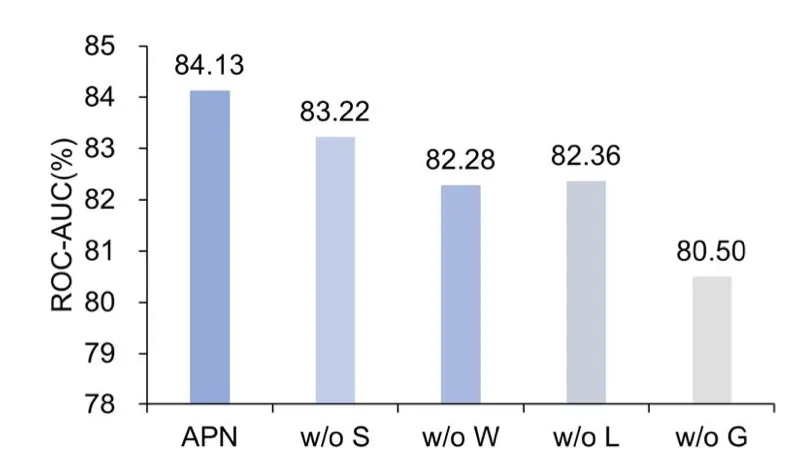 +
+*Figure 7* showcases the ablation study results on the Tox21 dataset, validating the effectiveness of different APN modules:
+
+1. **Global Attention Module**:
+ - Removing the attribute-guided global attention module (w/o G) significantly reduces performance, emphasizing its critical role in capturing relevant information for few-shot tasks.
+
+2. **Local Attention Module**:
+ - Removing the attribute-guided local attention module (w/o L) also decreases performance, though to a lesser extent than the global module, demonstrating its contribution to improved results.
+
+3. **Similarity and Weighted Prototypes**:
+ - Variants without dot-product similarity (w/o S) and weighted prototypes (w/o W) underperform compared to the complete APN model, underscoring the importance of integrating these components.
+
+These experiments reveal the synergistic effects of the global and local attention mechanisms and highlight the contribution of each module to overall performance improvement.
+
+---
+
+**Performance Enhancement with Three-Fingerprint Combinations**
+
+
+
+*Figure 7* showcases the ablation study results on the Tox21 dataset, validating the effectiveness of different APN modules:
+
+1. **Global Attention Module**:
+ - Removing the attribute-guided global attention module (w/o G) significantly reduces performance, emphasizing its critical role in capturing relevant information for few-shot tasks.
+
+2. **Local Attention Module**:
+ - Removing the attribute-guided local attention module (w/o L) also decreases performance, though to a lesser extent than the global module, demonstrating its contribution to improved results.
+
+3. **Similarity and Weighted Prototypes**:
+ - Variants without dot-product similarity (w/o S) and weighted prototypes (w/o W) underperform compared to the complete APN model, underscoring the importance of integrating these components.
+
+These experiments reveal the synergistic effects of the global and local attention mechanisms and highlight the contribution of each module to overall performance improvement.
+
+---
+
+**Performance Enhancement with Three-Fingerprint Combinations**
+
+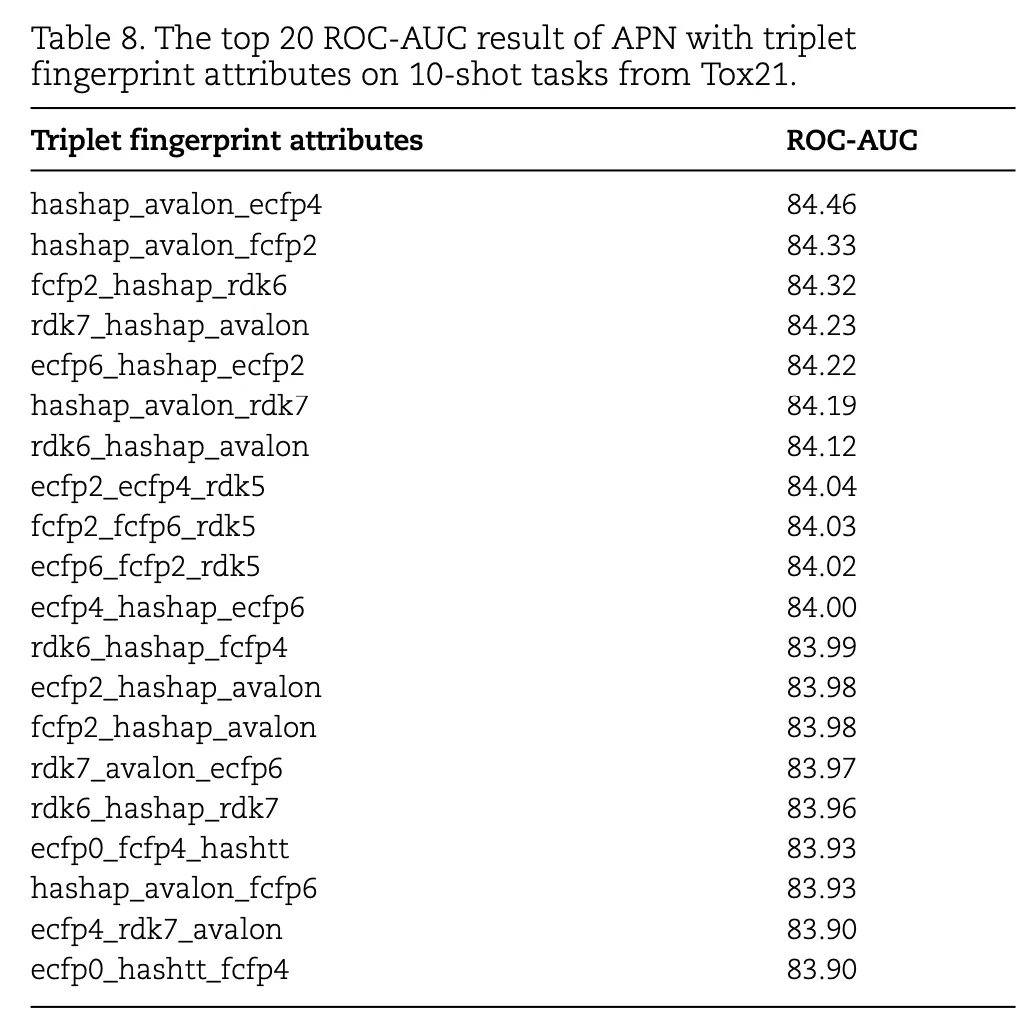 +
+*Table 8* lists the top 20 three-fingerprint combinations on the Tox21 dataset and their ROC-AUC scores:
+
+- The best combination, **hashap_avalon_ecfp4**, achieves a ROC-AUC of 84.46%, outperforming both single and double-fingerprint attributes.
+- Other effective combinations include **hashap_avalon_fcfp2** and **fcfp2_hashap_rdk6**, with scores of 84.33% and 84.32%, respectively, demonstrating the potential of combining multiple fingerprint attributes.
+
+These results validate that using three-fingerprint combinations significantly enhances prediction performance. Combinations involving attributes like HashAP and Avalon are particularly effective, showcasing the synergistic benefits of integrating multiple molecular fingerprints for few-shot molecular property prediction tasks.
+
+
+## Conclusions
+In this study, the authors proposed the Attribute-guided Prototype Network (APN), which integrates high-level molecular attributes with deep attributes generated through self-supervised learning to address the challenges of few-shot molecular property prediction (FS-MPP). Uni-Mol played a critical role in this model. As a self-supervised learning framework, Uni-Mol generates deep attributes that effectively capture the complex relationships between molecular structures and properties.
+
+By combining deep attributes generated by Uni-Mol (e.g., unimol_10conf) with traditional molecular fingerprint attributes, APN demonstrated outstanding performance across multiple datasets, including Tox21, SIDER, and MUV. The model significantly improved prediction accuracy, particularly in few-shot scenarios. Experimental results also revealed that APN, with its integration of local and global attention mechanisms, possesses strong generalization capabilities, making it highly effective for various molecular property prediction tasks.
+
+Future research could explore additional combinations of deep attributes and molecular fingerprints to further enhance the predictive power and applicability of FS-MPP, broadening its value in real-world applications.
+
+*Table 8* lists the top 20 three-fingerprint combinations on the Tox21 dataset and their ROC-AUC scores:
+
+- The best combination, **hashap_avalon_ecfp4**, achieves a ROC-AUC of 84.46%, outperforming both single and double-fingerprint attributes.
+- Other effective combinations include **hashap_avalon_fcfp2** and **fcfp2_hashap_rdk6**, with scores of 84.33% and 84.32%, respectively, demonstrating the potential of combining multiple fingerprint attributes.
+
+These results validate that using three-fingerprint combinations significantly enhances prediction performance. Combinations involving attributes like HashAP and Avalon are particularly effective, showcasing the synergistic benefits of integrating multiple molecular fingerprints for few-shot molecular property prediction tasks.
+
+
+## Conclusions
+In this study, the authors proposed the Attribute-guided Prototype Network (APN), which integrates high-level molecular attributes with deep attributes generated through self-supervised learning to address the challenges of few-shot molecular property prediction (FS-MPP). Uni-Mol played a critical role in this model. As a self-supervised learning framework, Uni-Mol generates deep attributes that effectively capture the complex relationships between molecular structures and properties.
+
+By combining deep attributes generated by Uni-Mol (e.g., unimol_10conf) with traditional molecular fingerprint attributes, APN demonstrated outstanding performance across multiple datasets, including Tox21, SIDER, and MUV. The model significantly improved prediction accuracy, particularly in few-shot scenarios. Experimental results also revealed that APN, with its integration of local and global attention mechanisms, possesses strong generalization capabilities, making it highly effective for various molecular property prediction tasks.
+
+Future research could explore additional combinations of deep attributes and molecular fingerprints to further enhance the predictive power and applicability of FS-MPP, broadening its value in real-world applications.