The RMSE of energy/atom and force increased with tamperatrue of AIMD #2252
-
|
(1) I used the AIMD method to simulate sulfide materials at 800 K, 1000 K, 1200 K, 1400 K and 1600 K using NVT ensemble for 80 ps, respectively. Then I trained the whole data to generate graph-compress.pb, and find the result is poor. So I trained the data at each temperature, and find that The RMSE of energy/atom and force increased with temperature. I reruned the AIMD at 1600 K for twice, the RMSE is also larger than that at lower temperautre. The AIMD parameters are all the same, excepts temperatures, and the training results differs. Is this phenomenon normal? How to understand this? The training steps are set to be only 20000. However, when I increased the training step to 100000, the result at 1600 K are still very poor, as saying “Garbage in, garbage out”. What should I do? (2) AIMD parameters are shown: PS: input.json is below: } |
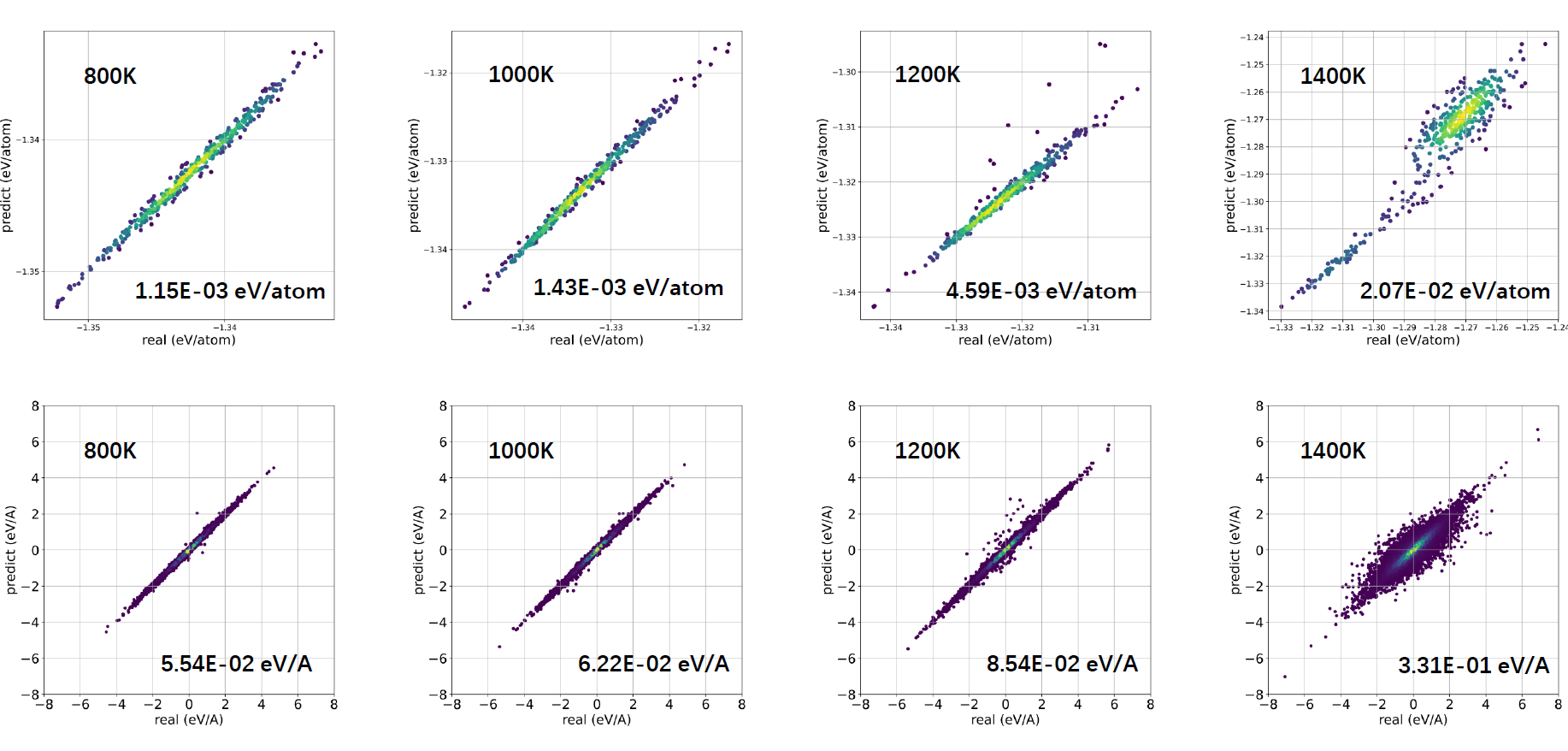
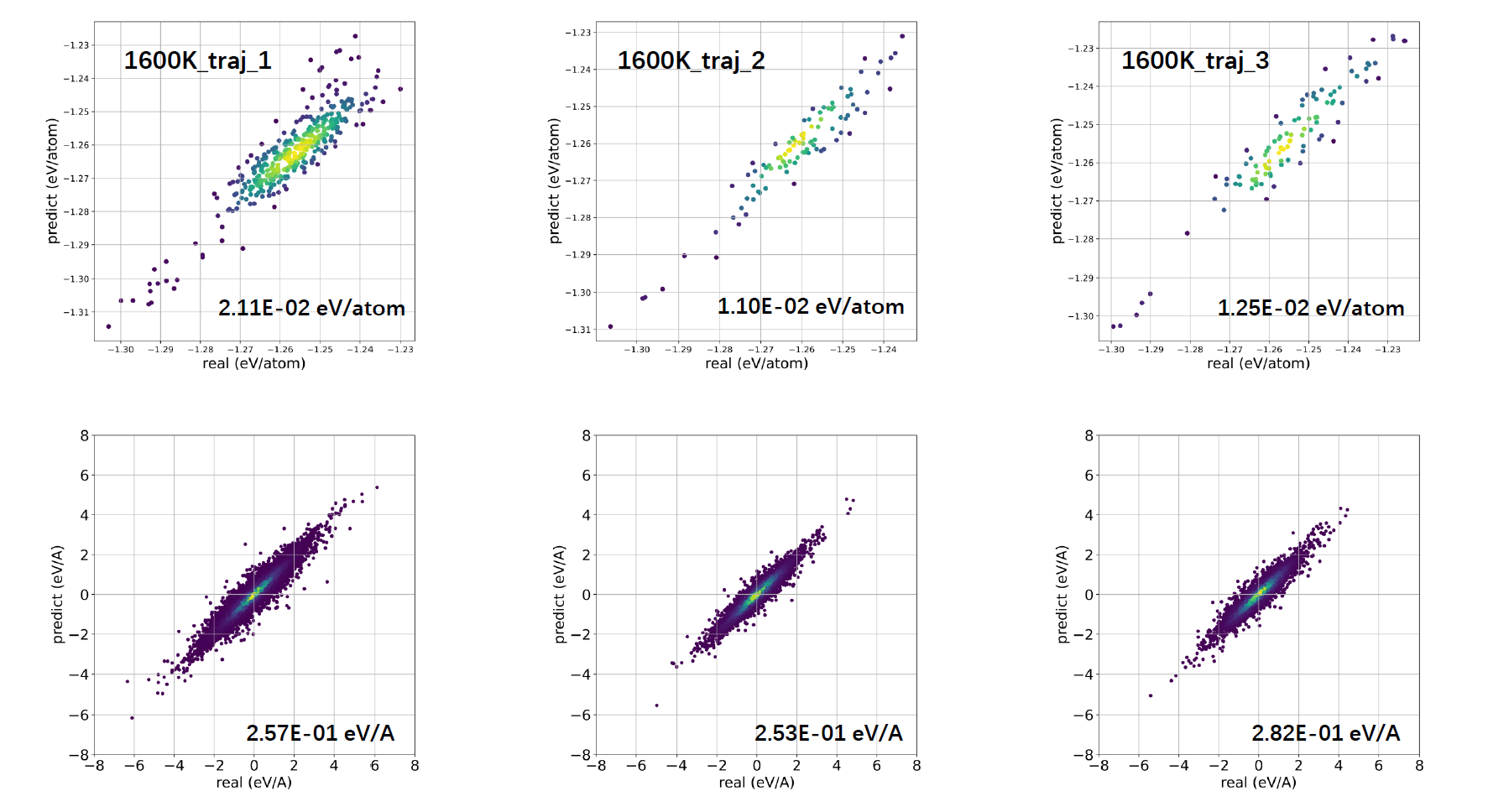
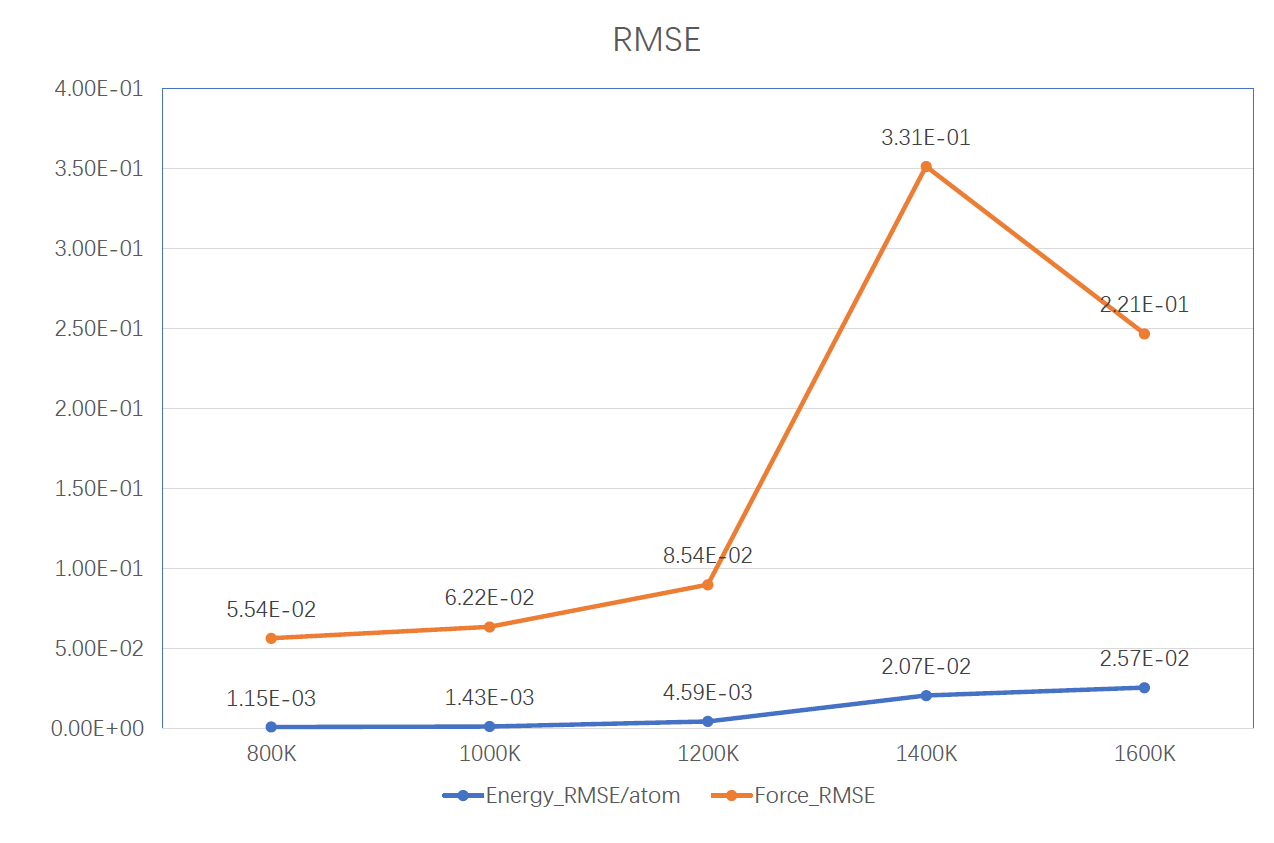
Beta Was this translation helpful? Give feedback.
Replies: 2 comments 4 replies
-
|
This behavior is similar to what I've seen using every MLIAP I've ever run. The configurational space sampled at 50 K is simply smaller than the configurational space sampled at 1600 K. As such, at 1600 K the potential is asked to predict the forces and energies of a more diverse set of structures, and will always have more difficulty doing so, whereas at 50 K the structures sampled in a MD run are going to be nearer to the ground state, and thus easier to predict. To an extent, you're just going to need a fairly large amount of data to allow for accurate interpolation at 1600 K. If you want to remedy this, you'll need a more diverse training set. You could run additional high(er) temperature ab initio molecular dynamics, and then add structures generated in that manner to the training set, or you could run an active learning-style algorithm, eg the DP-Gen [1] algorithm (listing here because its designed to interface with DeepMD-Kit) to generate a more diverse training set. If you want to go down the path of rerunning AIMD, I've generally seen doing a large number of short AIMD runs as preferable to running a few long AIMD runs in literature, though I'd recommend an active learning type approach, as you can generally fill in the gaps in the training set using less DFT time that way. Also, out of curiosity, what's the size both of the AIMD simulations (in number of atoms), and the number of structures you're training on. [1] Zhang, Yuzhi, et al. "DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models." Computer Physics Communications 253 (2020): 107206. I am not a dev of the project, and thus can't really comment deeply on the DeepMD-kit parameters used here. Though, in every sample training script I've seen for DeepMD-Kit, the max number of training steps is on the order of 10^6 rather than 2*10^4 as used here, so it could be that the potential isn't fully trained yet. In my own case, I still see improvement to the validation forces up to around 250k training steps. A useful metric for looking at this could be plotting the training loss (in lcurve.out) against the epoch number, and seeing if its still decreasing at the time training finishes. Edit, the cutoff seems fine; generally VASP wants 1.3-1.5x the max cutoff for running AIMD/Relaxation, and it appears you've satisfied that assuming you're using standard pseudopotentials, though definitely double check that. Depending on the size of the simulation, I might worry a tiny bit about the density of the KPOINT mesh, but I don't think it's the root of the problem here. |
Beta Was this translation helpful? Give feedback.
-
|
Thank you very much for your patient and meticulous answer, which has benefited me a lot. I re-trained data with 100k steps, and the RMSE decreased. I am still learning how to use DP-GEN, and the GPU servers won't be deployed until summer. Maybe I can try that way later. The ABC values are 10.86, 11.56 and 15.67 A, with alpha_beta_gama 90, 90, 90. The model has 64 atoms, and the AIMD is running via vasp_gam instead of vasp_std. I don't know what AIMD requires for the system size, and I don't know if such a setting is reasonable? Thanks again for your advice!!! |
Beta Was this translation helpful? Give feedback.
-
|
Based on the system size, and the vasp settings given, I doubt they are the source of the large error at high temperature. I'd definitely recommend training to a rather large total step size (1,000,000 is fairly common) to see if that can help you to further reduce error. The info linked in the FAQ in the other answer should also be helpful. |
Beta Was this translation helpful? Give feedback.
-
|
Yes, I'm runing a total step of 3,000,00, and it has not stopped. Hope to have a good results. |
Beta Was this translation helpful? Give feedback.
-
|
Please read FAQ:Why does a model have low precision:
|
Beta Was this translation helpful? Give feedback.
-
|
Thanks very much. I will read the FAQ carefully. BTW, happy Chinese new year. |
Beta Was this translation helpful? Give feedback.
This behavior is similar to what I've seen using every MLIAP I've ever run. The configurational space sampled at 50 K is simply smaller than the configurational space sampled at 1600 K. As such, at 1600 K the potential is asked to predict the forces and energies of a more diverse set of structures, and will always have more difficulty doing so, whereas at 50 K the structures sampled in a MD run are going to be nearer to the ground state, and thus easier to predict. To an extent, you're just going to need a fairly large amount of data to allow for accurate interpolation at 1600 K.
If you want to remedy this, you'll need a more diverse training set. You could run additional high(er) tempera…