Output from different fastq pairs mixing in final process #2336
-
|
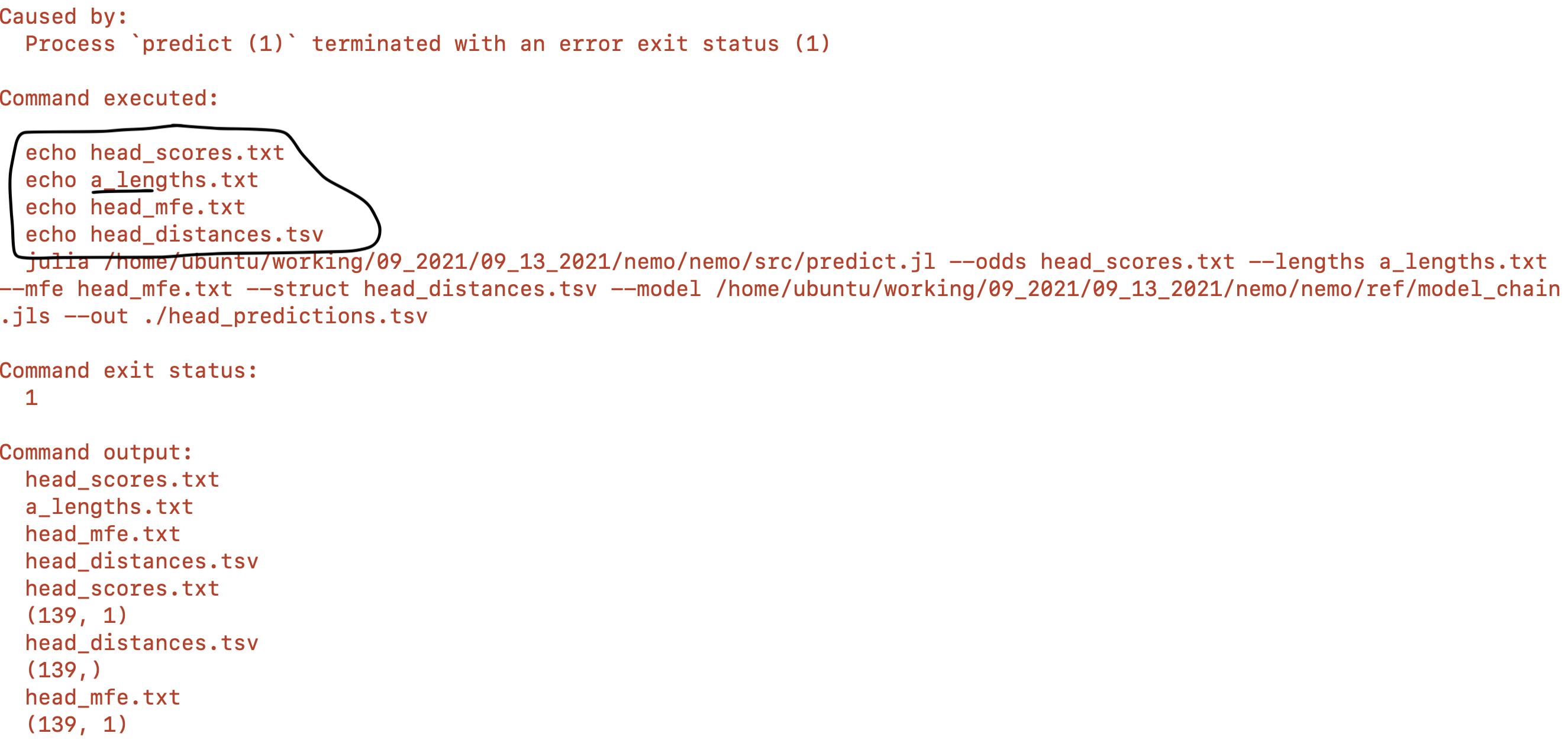
Hi everyone, I have a question for an issue I have not been able to resolve on my own. I wrote a pipeline to analyze RNAseq data. For each pair of FASTQ files, they are assembled and then passed into processes that do different analyses. The final step in the pipeline is taking 4 different outputs and passing them to a predictive model. What keeps happening to me however is that different pair_ids are being mixed together. I'm not really sure how this is possible. I have included a screen shot below. The general pattern is ${pair_id}_processname.tsv, however you can see where I underlined that an incorrect $pair_id is being passed into this process. If I run it again, the pair_ids seem essentially randomly shuffled, i.e. the names won't be the same every time and sometimes by luck it will run correctly. Anyone have any idea why this is happening? I'm sure I am doing something wrong...
|
Beta Was this translation helpful? Give feedback.
Replies: 3 comments 2 replies
-
|
A couple of comments here:
|
Beta Was this translation helpful? Give feedback.
-
|
Thanks, I've updated my code to reflect point 1 you made. For point 2: This would make a lot of sense. I'm testing the code on the first 100 lines and first 100000 lines of the fastq files, so this is very much the case. Is there a design pattern that would allow simultaneous execution of these different post-assembly processes, but still guarantee that they are collected by pair, rather than time to completion? Perhaps the |
Beta Was this translation helpful? Give feedback.
-
|
So, you have 4 channels, each of them with tuples of 2 elements, the pair ID is the first element for all of them. The first idea that pops in my mind is to mix the 4 channels into a single channel and then use groupTuple to obtain a tuple (pair id, [file from P1,...., file from P4]). There might be more elegant solutions, but this should work. |
Beta Was this translation helpful? Give feedback.
-
|
Thanks for your help, Anyway, here is the fix for anyone with similar issues: |
Beta Was this translation helpful? Give feedback.
-
|
My apologies, I could have sent you a small snippet with the channel manipulation I mentioned. But it's good you managed yourself anyway. You're on your way to be a groovy master! |
Beta Was this translation helpful? Give feedback.
Thanks for your help,
mixwas the answer but additional work was required. Unfortunately themixfunction does not emit in order and for this to work you need to know the order of the files. I don't know groovy at all so it took me many hours to figure out how to get the tuple (pair_id, [file 1, file 2, ... file N]) in order. The fix ended up being quite elegant, but thetoSortedfunction is presumably found in the groovy documentation, and is no where in the nextflow api. The other key realization was that the file paths which appear as plain strings when you .view() turn out to have a.nameattribute, which allows you to sort by the base name of the file. Otherwise it would be impossibl…