diff --git a/.github/workflows/nf-test.yml b/.github/workflows/nf-test.yml
index e20bf6d..8886f92 100644
--- a/.github/workflows/nf-test.yml
+++ b/.github/workflows/nf-test.yml
@@ -64,11 +64,12 @@ jobs:
runs-on: # use self-hosted runners
- runs-on=${{ github.run_id }}-nf-test
- runner=4cpu-linux-x64
+ - volume=80gb
strategy:
fail-fast: false
matrix:
shard: ${{ fromJson(needs.nf-test-changes.outputs.shard) }}
- profile: [conda, docker, singularity]
+ profile: [docker, singularity] # TODO: add conda back, but only for cellpose
isMain:
- ${{ github.base_ref == 'master' || github.base_ref == 'main' }}
# Exclude conda and singularity on dev
diff --git a/.gitignore b/.gitignore
index a42ce01..d75d93b 100644
--- a/.gitignore
+++ b/.gitignore
@@ -7,3 +7,7 @@ testing/
testing*
*.pyc
null/
+sandbox
+samplesheets
+lint_*
+.nf-test*
diff --git a/CHANGELOG.md b/CHANGELOG.md
index d7617f1..9f2d459 100644
--- a/CHANGELOG.md
+++ b/CHANGELOG.md
@@ -9,8 +9,4 @@ Initial release of nf-core/sopa, created with the [nf-core](https://nf-co.re/) t
### `Added`
-### `Fixed`
-
-### `Dependencies`
-
-### `Deprecated`
+Sopa can be run with all the technologies currently supported - including Visium HD.
diff --git a/CITATIONS.md b/CITATIONS.md
index 3a62896..2f2372f 100644
--- a/CITATIONS.md
+++ b/CITATIONS.md
@@ -1,5 +1,9 @@
# nf-core/sopa: Citations
+## [sopa](https://www.nature.com/articles/s41467-024-48981-z)
+
+> Blampey, Q., Mulder, K., Gardet, M. et al. Sopa: a technology-invariant pipeline for analyses of image-based spatial omics. Nat Commun 15, 4981 (2024). https://doi.org/10.1038/s41467-024-48981-z
+
## [nf-core](https://pubmed.ncbi.nlm.nih.gov/32055031/)
> Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020 Mar;38(3):276-278. doi: 10.1038/s41587-020-0439-x. PubMed PMID: 32055031.
@@ -10,6 +14,22 @@
## Pipeline tools
+- [AnnData](https://github.com/scverse/anndata)
+
+ > Virshup I, Rybakov S, Theis FJ, Angerer P, Wolf FA. bioRxiv 2021.12.16.473007; doi: https://doi.org/10.1101/2021.12.16.473007
+
+- [Scanpy](https://github.com/theislab/scanpy)
+
+ > Wolf F, Angerer P, Theis F. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15 (2018). doi: https://doi.org/10.1186/s13059-017-1382-0
+
+- [Space Ranger](https://www.10xgenomics.com/support/software/space-ranger)
+
+ > 10x Genomics Space Ranger 2.1.0 [Online]
+
+- [SpatialData](https://www.biorxiv.org/content/10.1101/2023.05.05.539647v1)
+
+ > Marconato L, Palla G, Yamauchi K, Virshup I, Heidari E, Treis T, Toth M, Shrestha R, Vöhringer H, Huber W, Gerstung M, Moore J, Theis F, Stegle O. SpatialData: an open and universal data framework for spatial omics. bioRxiv 2023.05.05.539647; doi: https://doi.org/10.1101/2023.05.05.539647
+
## Software packaging/containerisation tools
- [Anaconda](https://anaconda.com)
diff --git a/README.md b/README.md
index 254dbc5..77035c1 100644
--- a/README.md
+++ b/README.md
@@ -21,47 +21,52 @@
## Introduction
-**nf-core/sopa** is a bioinformatics pipeline that ...
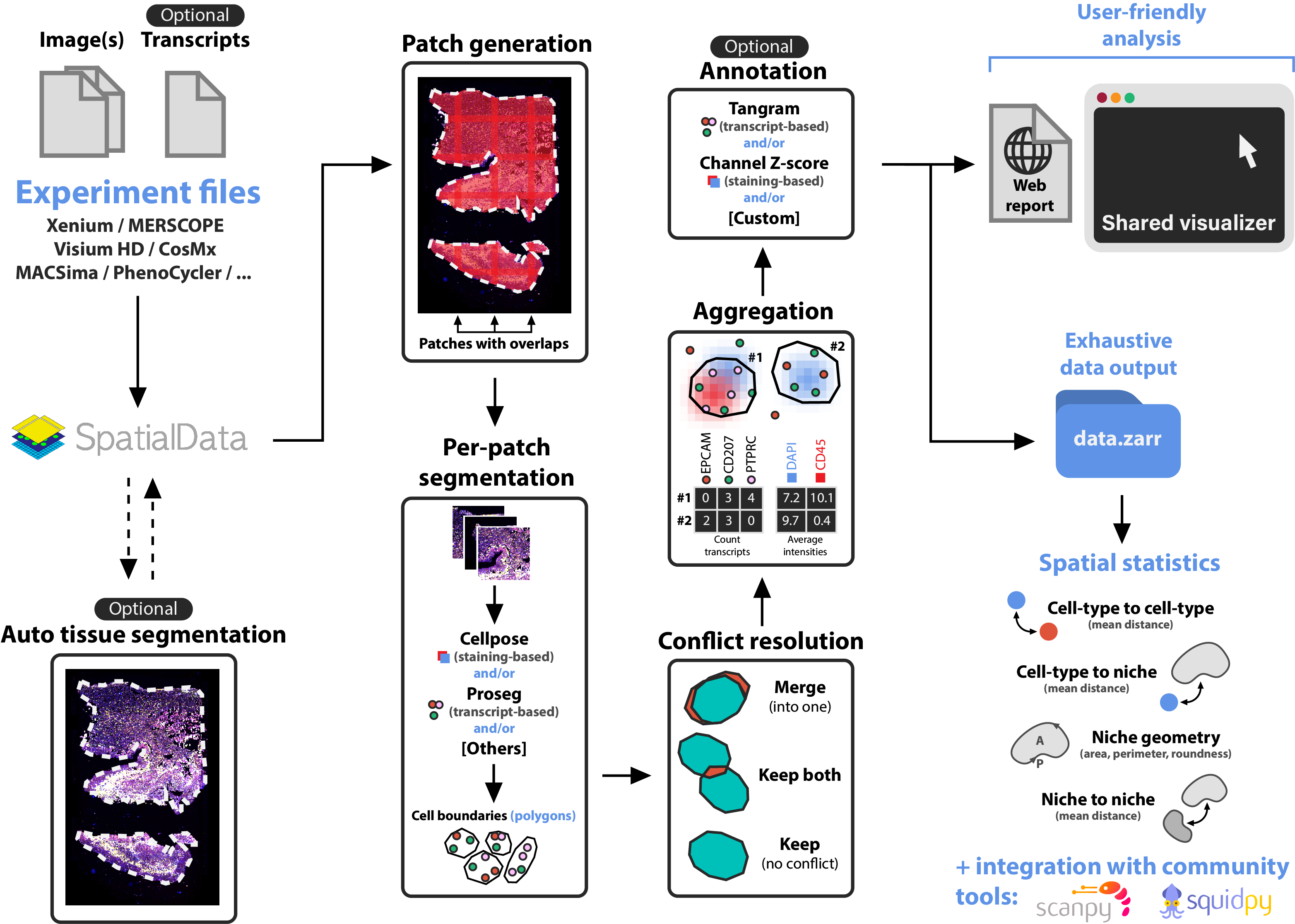
+**nf-core/sopa** is the Nextflow version of [Sopa](https://github.com/gustaveroussy/sopa). Built on top of [SpatialData](https://github.com/scverse/spatialdata), Sopa enables processing and analyses of spatial omics data with single-cell resolution (spatial transcriptomics or multiplex imaging data) using a standard data structure and output. We currently support the following technologies: Xenium, Visium HD, MERSCOPE, CosMX, PhenoCycler, MACSima, Molecural Cartography, and others. It outputs a `.zarr` directory containing a processed [SpatialData](https://github.com/scverse/spatialdata) object, and a `.explorer` directory for visualization.
-
+> [!WARNING]
+> If you are interested in the main Sopa python package, refer to [this Sopa repository](https://github.com/gustaveroussy/sopa). Else, if you want to use Nextflow, you are in the good place.
+
+
+  +
+
+
+1. (Visium HD only) Raw data processing with Space Ranger
+2. (Optional) Tissue segmentation
+3. Cell segmentation with Cellpose, Baysor, Proseg, Comseg, Stardist, ...
+4. Aggregation, i.e. counting the transcripts inside the cells and/or averaging the channel intensities inside cells
+5. (Optional) Cell-type annotation
+6. User-friendly output creation for visualization and quick analysis
+7. Full [SpatialData](https://github.com/scverse/spatialdata) object export as a `.zarr` directory
-
-
+After running `nf-core/sopa`, you can continue analyzing your `SpatialData` object with [`sopa` as a Python package](https://github.com/gustaveroussy/sopa).
## Usage
> [!NOTE]
> If you are new to Nextflow and nf-core, please refer to [this page](https://nf-co.re/docs/usage/installation) on how to set-up Nextflow. Make sure to [test your setup](https://nf-co.re/docs/usage/introduction#how-to-run-a-pipeline) with `-profile test` before running the workflow on actual data.
-
+Then, choose the Sopa parameters. You can find existing Sopa params files [here](https://github.com/gustaveroussy/sopa/tree/main/workflow/config), and follow the [corresponding README instructions](https://github.com/gustaveroussy/sopa/blob/main/workflow/config/README.md) of to get your `-params-file` argument.
Now, you can run the pipeline using:
-
-
```bash
nextflow run nf-core/sopa \
-profile \
--input samplesheet.csv \
+ -params-file \
--outdir
```
@@ -78,11 +83,12 @@ For more details about the output files and reports, please refer to the
## Credits
-nf-core/sopa was originally written by Quentin Blampey.
+nf-core/sopa was originally written by [Quentin Blampey](https://github.com/quentinblampey) during his work at the following institutions: CentraleSupélec, Gustave Roussy Institute, Université Paris-Saclay, and Cure51.
We thank the following people for their extensive assistance in the development of this pipeline:
-
+- [Matthias Hörtenhuber](https://github.com/mashehu)
+- [Kevin Weiss](https://github.com/kweisscure51)
## Contributions and Support
@@ -95,10 +101,16 @@ For further information or help, don't hesitate to get in touch on the [Slack `#
-
-
An extensive list of references for the tools used by the pipeline can be found in the [`CITATIONS.md`](CITATIONS.md) file.
+You can cite the `sopa` publication as follows:
+
+> Sopa: a technology-invariant pipeline for analyses of image-based spatial omics.
+>
+> Quentin Blampey, Kevin Mulder, Margaux Gardet, Stergios Christodoulidis, Charles-Antoine Dutertre, Fabrice André, Florent Ginhoux & Paul-Henry Cournède.
+>
+> _Nat Commun._ 2024 June 11. doi: [10.1038/s41467-024-48981-z](https://doi.org/10.1038/s41467-024-48981-z)
+
You can cite the `nf-core` publication as follows:
> **The nf-core framework for community-curated bioinformatics pipelines.**
diff --git a/assets/schema_input.json b/assets/schema_input.json
index 542fd19..6f93309 100644
--- a/assets/schema_input.json
+++ b/assets/schema_input.json
@@ -10,24 +10,90 @@
"sample": {
"type": "string",
"pattern": "^\\S+$",
- "errorMessage": "Sample name must be provided and cannot contain spaces",
+ "errorMessage": "Sample name cannot contain spaces",
+ "meta": ["sample"]
+ },
+ "id": {
+ "type": "string",
+ "pattern": "^\\S+$",
+ "errorMessage": "ID cannot contain spaces",
"meta": ["id"]
},
- "fastq_1": {
+ "data_path": {
+ "type": "string",
+ "pattern": "^\\S+$",
+ "format": "path",
+ "exists": true,
+ "errorMessage": "Data path must exist and not contain spaces"
+ },
+ "fastq_dir": {
+ "type": "string",
+ "pattern": "^\\S+$",
+ "format": "path",
+ "exists": true,
+ "errorMessage": "Path to the fastq directory must exist and not contain spaces",
+ "meta": ["fastq_dir"]
+ },
+ "cytaimage": {
+ "type": "string",
+ "pattern": "^\\S+(tif|tiff)$",
+ "format": "path",
+ "exists": true,
+ "errorMessage": "Path to the cytaimage file must exist, not contain spaces, and be a .tif or .tiff file",
+ "meta": ["cytaimage"]
+ },
+ "colorizedimage": {
+ "type": "string",
+ "pattern": "^\\S+(tif|tiff|jpg|jpeg|btf)$",
+ "format": "path",
+ "exists": true,
+ "errorMessage": "Path to the colorizedimage file must exist, not contain spaces, and be a .tif, .tiff, .btf, .jpg or .jpeg file",
+ "meta": ["colorizedimage"]
+ },
+ "darkimage": {
+ "type": "string",
+ "pattern": "^\\S+(tif|tiff|jpg|jpeg|btf)$",
+ "format": "path",
+ "exists": true,
+ "errorMessage": "Path to the darkimage file must exist, not contain spaces, and be a .tif, .tiff, .btf, .jpg or .jpeg file",
+ "meta": ["darkimage"]
+ },
+ "image": {
+ "type": "string",
+ "pattern": "^\\S+(tif|tiff|jpg|jpeg|btf)$",
+ "format": "path",
+ "exists": true,
+ "errorMessage": "Path to the image must exist, not contain spaces, and be a .tif, .tiff, .btf, .jpg or .jpeg file",
+ "meta": ["image"]
+ },
+ "slide": {
+ "type": "string",
+ "pattern": "^\\S+$",
+ "errorMessage": "Slide name cannot contain spaces",
+ "meta": ["slide"]
+ },
+ "area": {
+ "type": "string",
+ "pattern": "^\\S+$",
+ "errorMessage": "Area name cannot contain spaces",
+ "meta": ["area"]
+ },
+ "manual_alignment": {
"type": "string",
- "format": "file-path",
+ "pattern": "^\\S+json$",
+ "format": "path",
"exists": true,
- "pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
- "errorMessage": "FastQ file for reads 1 must be provided, cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
+ "errorMessage": "Path to the manual_alignment must exist, not contain spaces, and be a .json file",
+ "meta": ["manual_alignment"]
},
- "fastq_2": {
+ "slidefile": {
"type": "string",
- "format": "file-path",
+ "pattern": "^\\S+json$",
+ "format": "path",
"exists": true,
- "pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
- "errorMessage": "FastQ file for reads 2 cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
+ "errorMessage": "Path to the slidefile must exist, not contain spaces, and be a .json file",
+ "meta": ["slidefile"]
}
- },
- "required": ["sample", "fastq_1"]
+ }
}
}
diff --git a/conf/base.config b/conf/base.config
index 2431ea1..53ee460 100644
--- a/conf/base.config
+++ b/conf/base.config
@@ -9,8 +9,6 @@
*/
process {
-
- // TODO nf-core: Check the defaults for all processes
cpus = { 1 * task.attempt }
memory = { 6.GB * task.attempt }
time = { 4.h * task.attempt }
@@ -19,13 +17,6 @@ process {
maxRetries = 1
maxErrors = '-1'
- // Process-specific resource requirements
- // NOTE - Please try and reuse the labels below as much as possible.
- // These labels are used and recognised by default in DSL2 files hosted on nf-core/modules.
- // If possible, it would be nice to keep the same label naming convention when
- // adding in your local modules too.
- // TODO nf-core: Customise requirements for specific processes.
- // See https://www.nextflow.io/docs/latest/config.html#config-process-selectors
withLabel:process_single {
cpus = { 1 }
memory = { 6.GB * task.attempt }
@@ -52,6 +43,11 @@ process {
withLabel:process_high_memory {
memory = { 200.GB * task.attempt }
}
+ withName:PATCH_SEGMENTATION_PROSEG {
+ cpus = { 8 * task.attempt }
+ memory = { 200.GB * task.attempt }
+ time = { 10.d * task.attempt }
+ }
withLabel:error_ignore {
errorStrategy = 'ignore'
}
diff --git a/conf/modules.config b/conf/modules.config
index e27fd28..6cb190b 100644
--- a/conf/modules.config
+++ b/conf/modules.config
@@ -11,11 +11,12 @@
*/
process {
-
- publishDir = [
- path: { "${params.outdir}/${task.process.tokenize(':')[-1].tokenize('_')[0].toLowerCase()}" },
- mode: params.publish_dir_mode,
- saveAs: { filename -> filename.equals('versions.yml') ? null : filename }
- ]
-
+ withName: SPACERANGER_COUNT {
+ ext.args = '--create-bam="false"'
+ publishDir = [
+ path: { "${params.outdir}/${meta.sample}_spaceranger" },
+ mode: params.publish_dir_mode,
+ saveAs: { filename -> filename.equals('versions.yml') ? null : filename },
+ ]
+ }
}
diff --git a/conf/test.config b/conf/test.config
index 8d9a94b..0befa9f 100644
--- a/conf/test.config
+++ b/conf/test.config
@@ -14,16 +14,53 @@ process {
resourceLimits = [
cpus: 4,
memory: '15.GB',
- time: '1.h'
+ time: '1.h',
]
}
params {
- config_profile_name = 'Test profile'
+ config_profile_name = 'Test profile with Proseg'
config_profile_description = 'Minimal test dataset to check pipeline function'
// Input data
- // TODO nf-core: Specify the paths to your test data on nf-core/test-datasets
- // TODO nf-core: Give any required params for the test so that command line flags are not needed
- input = params.pipelines_testdata_base_path + 'viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv'
+ input = "${baseDir}/tests/samplesheet.csv"
+
+ read = [
+ technology: "toy_dataset",
+ kwargs: [
+ genes: 500
+ ],
+ ]
+
+ patchify = [
+ patch_width_microns: 400,
+ patch_overlap_microns: 20,
+ ]
+
+ segmentation.proseg.prior_shapes_key = "auto"
+
+ aggregate = [
+ aggregate_channels: true,
+ min_transcripts: 5,
+ ]
+
+ annotation = [
+ method: "fluorescence",
+ args: [
+ marker_cell_dict: [
+ CK: "Tumoral cell",
+ CD3: "T cell",
+ CD20: "B cell",
+ ]
+ ],
+ ]
+
+ scanpy_preprocess = [
+ check_counts: false
+ ]
+
+ explorer = [
+ ram_threshold_gb: 4,
+ pixel_size: 0.1,
+ ]
}
diff --git a/conf/test_baysor.config b/conf/test_baysor.config
new file mode 100644
index 0000000..0f4a3b9
--- /dev/null
+++ b/conf/test_baysor.config
@@ -0,0 +1,74 @@
+/*
+~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
+ Nextflow config file for running minimal tests
+~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
+ Defines input files and everything required to run a fast and simple pipeline test.
+
+ Use as follows:
+ nextflow run nf-core/sopa -profile test_baysor, --outdir
+
+----------------------------------------------------------------------------------------
+*/
+
+process {
+ resourceLimits = [
+ cpus: 4,
+ memory: '15.GB',
+ time: '1.h',
+ ]
+}
+
+params {
+ config_profile_name = 'Test profile'
+ config_profile_description = 'Minimal test dataset to check pipeline function'
+
+ // Input data
+ input = "${baseDir}/tests/samplesheet.csv"
+
+ read.technology = "toy_dataset"
+
+ patchify = [
+ patch_width_microns: 400,
+ patch_overlap_microns: 20,
+ ]
+
+ segmentation.baysor = [
+ min_area: 10,
+ config: [
+ data: [
+ force_2d: true,
+ min_molecules_per_cell: 10,
+ x: "x",
+ y: "y",
+ z: "z",
+ gene: "genes",
+ min_molecules_per_gene: 0,
+ min_molecules_per_segment: 3,
+ confidence_nn_id: 6,
+ ],
+ segmentation: [
+ scale: 3,
+ scale_std: "25%",
+ prior_segmentation_confidence: 0,
+ ],
+ ],
+ ]
+
+ aggregate = [
+ aggregate_channels: true,
+ min_transcripts: 5,
+ ]
+
+ annotation = [
+ method: "tangram",
+ args: [
+ sc_reference_path: "https://github.com/gustaveroussy/sopa/raw/refs/heads/main/tests/toy_tangram_ref.h5ad",
+ cell_type_key: "ct",
+ ],
+ ]
+
+ explorer = [
+ ram_threshold_gb: 4,
+ pixel_size: 0.1,
+ ]
+}
diff --git a/conf/test_cellpose.config b/conf/test_cellpose.config
new file mode 100644
index 0000000..0351f7e
--- /dev/null
+++ b/conf/test_cellpose.config
@@ -0,0 +1,49 @@
+/*
+~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
+ Nextflow config file for running minimal tests
+~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
+ Defines input files and everything required to run a fast and simple pipeline test.
+
+ Use as follows:
+ nextflow run nf-core/sopa -profile test_cellpose, --outdir
+
+----------------------------------------------------------------------------------------
+*/
+
+process {
+ resourceLimits = [
+ cpus: 4,
+ memory: '15.GB',
+ time: '1.h',
+ ]
+}
+
+params {
+ config_profile_name = 'Test profile with Cellpose'
+ config_profile_description = 'Minimal test dataset to check pipeline function'
+
+ // Input data
+ input = "${baseDir}/tests/samplesheet.csv"
+
+ read.technology = "toy_dataset"
+
+ patchify = [
+ patch_width_pixel: 5000,
+ patch_overlap_pixel: 50,
+ ]
+
+ segmentation.cellpose = [
+ diameter: 35,
+ channels: ["DAPI"],
+ flow_threshold: 2,
+ cellprob_threshold: -6,
+ min_area: 2500,
+ ]
+
+ aggregate.aggregate_channels = true
+
+ explorer = [
+ ram_threshold_gb: 4,
+ pixel_size: 0.1,
+ ]
+}
diff --git a/conf/test_comseg.config b/conf/test_comseg.config
new file mode 100644
index 0000000..e79d265
--- /dev/null
+++ b/conf/test_comseg.config
@@ -0,0 +1,64 @@
+/*
+~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
+ Nextflow config file for running minimal tests
+~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
+ Defines input files and everything required to run a fast and simple pipeline test.
+
+ Use as follows:
+ nextflow run nf-core/sopa -profile test_comseg, --outdir
+
+----------------------------------------------------------------------------------------
+*/
+
+process {
+ resourceLimits = [
+ cpus: 4,
+ memory: '15.GB',
+ time: '1.h',
+ ]
+}
+
+params {
+ config_profile_name = 'Test profile with Comseg'
+ config_profile_description = 'Minimal test dataset to check pipeline function'
+
+ // Input data
+ input = "${baseDir}/tests/samplesheet.csv"
+
+ read.technology = "toy_dataset"
+
+
+ patchify = [
+ patch_width_microns: 400,
+ patch_overlap_microns: 20,
+ ]
+
+ segmentation.comseg = [
+ min_area: 10,
+ prior_shapes_key: "cells",
+ config: [
+ dict_scale: [
+ x: 1,
+ y: 1,
+ z: 1,
+ ],
+ mean_cell_diameter: 15,
+ max_cell_radius: 25,
+ allow_disconnected_polygon: false,
+ alpha: 0.5,
+ min_rna_per_cell: 5,
+ gene_column: "genes",
+ norm_vector: false,
+ ],
+ ]
+
+ aggregate = [
+ aggregate_channels: true,
+ min_transcripts: 5,
+ ]
+
+ explorer = [
+ ram_threshold_gb: 4,
+ pixel_size: 0.1,
+ ]
+}
diff --git a/conf/test_full.config b/conf/test_full.config
index 63eed15..509e3ca 100644
--- a/conf/test_full.config
+++ b/conf/test_full.config
@@ -11,14 +11,34 @@
*/
params {
- config_profile_name = 'Full test profile'
+ config_profile_name = 'Full test profile'
config_profile_description = 'Full test dataset to check pipeline function'
- // Input data for full size test
- // TODO nf-core: Specify the paths to your full test data ( on nf-core/test-datasets or directly in repositories, e.g. SRA)
- // TODO nf-core: Give any required params for the test so that command line flags are not needed
- input = params.pipelines_testdata_base_path + 'viralrecon/samplesheet/samplesheet_full_illumina_amplicon.csv'
+ // Input data
+ input = "${baseDir}/tests/samplesheet.csv"
- // Fasta references
- fasta = params.pipelines_testdata_base_path + 'viralrecon/genome/NC_045512.2/GCF_009858895.2_ASM985889v3_genomic.200409.fna.gz'
+ read = [
+ technology: "toy_dataset",
+ kwargs: [
+ genes: 500,
+ length: 10000,
+ ],
+ ]
+
+ patchify = [
+ patch_width_microns: 400,
+ patch_overlap_microns: 20,
+ ]
+
+ segmentation.proseg.prior_shapes_key = "auto"
+
+ aggregate = [
+ aggregate_channels: true,
+ min_transcripts: 5,
+ ]
+
+ explorer = [
+ ram_threshold_gb: 4,
+ pixel_size: 0.1,
+ ]
}
diff --git a/docs/output.md b/docs/output.md
index 8aa16f1..bcf45e3 100644
--- a/docs/output.md
+++ b/docs/output.md
@@ -6,14 +6,48 @@ This document describes the output produced by the pipeline.
The directories listed below will be created in the results directory after the pipeline has finished. All paths are relative to the top-level results directory.
-
-
## Pipeline overview
-The pipeline is built using [Nextflow](https://www.nextflow.io/) and processes data using the following steps:
+The pipeline is built using [Nextflow](https://www.nextflow.io/) and outputs the following information:
+- [SpatialData directory](#spatialdata-directory) - Full [SpatialData](https://spatialdata.scverse.org/en/stable/) object with the segmented and aggregated data.
+- [Explorer directory](#explorer-directory) - Visualization and quick analysis directory
+- [VisiumHD-specific outputs](#visiumhd-specific-outputs) - Outputs of Space Ranger
- [Pipeline information](#pipeline-information) - Report metrics generated during the workflow execution
+### SpatialData directory
+
+
+Output files
+
+- `{sample}.zarr/`
+ - Spatial elements: `images/`, `shapes/`, `tables/`, `points/`, ...
+
+
+
+The `{sample}.zarr` directory contains a [SpatialData](https://spatialdata.scverse.org/en/stable/) object, where the `sample` name is either (i) specified by the samplesheet, or (ii) based on the name of the corresponding input directory.
+
+Refer to the [SpatialData docs](https://spatialdata.scverse.org/en/stable/) for usage details, or to the [documentation of `sopa` as a Python package](https://gustaveroussy.github.io/sopa/). If you are not familiar with `SpatialData`, you can also use directly the extracted `AnnData` object (see below).
+
+### Explorer directory
+
+
+Output files
+
+- `{sample}.explorer/`
+ - Sopa quality controls: `report.html`
+ - AnnData object (extracted from the above SpatialData object): `adata.h5ad`
+ - Xenium Explorer file: `experiment.xenium`. Double-click on it to open it on the Xenium Explorer; you can download the software [here](https://www.10xgenomics.com/support/software/xenium-explorer/downloads).
+ - Other files related and required by the Xenium Explorer.
+
+
+
+The `{sample}.explorer` directory can be used for visualization and quick analysis.
+
+### VisiumHD-specific outputs
+
+**(Only for Visium HD)** a `{sample}_spaceranger/outs` directory with the outputs of Space Ranger. See [the official 10X Genomics documentation](https://www.10xgenomics.com/support/software/space-ranger/latest/analysis/outputs/output-overview) for more details.
+
### Pipeline information
diff --git a/docs/usage.md b/docs/usage.md
index 2fdb33f..91186a6 100644
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -4,62 +4,106 @@
> _Documentation of pipeline parameters is generated automatically from the pipeline schema and can no longer be found in markdown files._
-## Introduction
-
-
-
## Samplesheet input
-You will need to create a samplesheet with information about the samples you would like to analyse before running the pipeline. Use this parameter to specify its location. It has to be a comma-separated file with 3 columns, and a header row as shown in the examples below.
+You will need to create a samplesheet with information about the samples you would like to analyse before running the pipeline. Use this parameter to specify its location. It has to be a comma-separated file with 2 columns, and a header row as shown in the examples below.
```bash
--input '[path to samplesheet file]'
```
-### Multiple runs of the same sample
+### Main technologies
-The `sample` identifiers have to be the same when you have re-sequenced the same sample more than once e.g. to increase sequencing depth. The pipeline will concatenate the raw reads before performing any downstream analysis. Below is an example for the same sample sequenced across 3 lanes:
+For all technologies supported by Sopa, the samplesheet lists the `data_path` to each sample data directory, and optionally a `sample` column to choose the name of the output directories.
-```csv title="samplesheet.csv"
-sample,fastq_1,fastq_2
-CONTROL_REP1,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz
-CONTROL_REP1,AEG588A1_S1_L003_R1_001.fastq.gz,AEG588A1_S1_L003_R2_001.fastq.gz
-CONTROL_REP1,AEG588A1_S1_L004_R1_001.fastq.gz,AEG588A1_S1_L004_R2_001.fastq.gz
-```
+> [!NOTE]
+> For **Visium HD only**, the samplesheet is different, please refer to the next section instead.
-### Full samplesheet
+The concerned technologies are: `xenium`, `merscope`, `cosmx`, `molecular_cartography`, `macsima`, `phenocycler`, `ome_tif`, and `hyperion`.
-The pipeline will auto-detect whether a sample is single- or paired-end using the information provided in the samplesheet. The samplesheet can have as many columns as you desire, however, there is a strict requirement for the first 3 columns to match those defined in the table below.
+| Column | Description |
+| ----------- | ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
+| `data_path` | **Path to the raw data**; a directory containing the output of the machine with the data of a single sample or region. Typically, this directory contains one or multiple image(s), and a transcript file (`.csv` or `.parquet`) for transcriptomics technologies. See more details below. _Required_ |
+| `sample` | **Custom sample ID (optional)**; designates the sample ID; must be unique for each patient. It will be used in the output directories names: `{sample}.zarr` and `{sample}.explorer`. _Optional, Default: the basename of `data_path` (i.e., the last directory component of `data_path`)_ |
+
+Here is a samplesheet example for two samples:
+
+`samplesheet.csv`:
+
+```csv title="samplesheet.csv"
+sample,data_path
+SAMPLE1,/path/to/one/merscope_directory
+SAMPLE2,/path/to/another/merscope_directory
+```
-A final samplesheet file consisting of both single- and paired-end data may look something like the one below. This is for 6 samples, where `TREATMENT_REP3` has been sequenced twice.
+We also provide a detailed description of what `data_path` should contain, depending on the technologies:
+
+| Technology | `data_path` directory content |
+| --------------------- | ---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
+| xenium | `transcripts.parquet`, `experiment.xenium`, and `morphology_focus.ome.tif` or a morphology directory. |
+| merscope | `detected_transcripts.csv`, all the images under the `images` subdirectory, and `images/micron_to_mosaic_pixel_transform.csv` (affine transformation) |
+| cosmx | `*_fov_positions_file.csv` or `*_fov_positions_file.csv.gz` (FOV locations),`Morphology2D` (directory with all the FOVs morphology images), and `*_tx_file.csv.gz` or `*_tx_file.csv` (transcripts location and names) |
+| molecular_cartography | Multiple `.tiff` images and `_results.txt` files. |
+| macsima | Multiple `.tif` images |
+| phenocycler | For this technology, `data_path` is not a directory, but a `.qptiff` or `.tif` file containing all channels for a given sample. |
+| hyperion | Multiple `.tif` images |
+| ome_tif | Generic reader for which `data_path` is not a directory, but a `.ome.tif` file containing all channels for a given sample. |
+
+### Visium HD
+
+Some extra columns need to be provided specifically for Visium HD. This is because we need to run [Space Ranger](https://www.10xgenomics.com/support/software/space-ranger/latest) before running Sopa. Note that the `image` is the full-resolution microscopy image (not the cytassist image) and is **required** by Sopa as we'll run cell segmentation on the H&E full-resolution slide. For more details, see the [`spaceranger-count` arguments](https://nf-co.re/modules/spaceranger_count).
+
+| Column | Description |
+| ------------------ | -------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
+| `sample` | **Sample ID name**; designates the sample ID; must be unique for each slide. It will be used in the output directories names: `{sample}.zarr` and `{sample}.explorer`. _Required_ |
+| `id` | Name of the slide to be provided to Space Ranger. The sample can be deduced from the fastq*dir, as the fastq files should have the format `\_S\_L001*_001.fastq.gz`(where N is a number, and XX can be R1, R2, I1 or I2). By default, use the`sample` name. \_Optional_ |
+| `fastq_dir` | Path to directory where the sample FASTQ files are stored. May be a `.tar.gz` file instead of a directory. _Required_ |
+| `image` | Brightfield microscopy image. _Required_ |
+| `cytaimage` | Brightfield tissue image captured with Cytassist device. _Required_ |
+| `slide` | The Visium slide ID used for the sequencing. _Required_ |
+| `area` | Which slide area contains the tissue sample. _Required_ |
+| `manual_alignment` | Path to the manual alignment file. _Optional_ |
+| `slidefile` | Slide specification as JSON. Overrides `slide` and `area` if specified. _Optional_ |
+| `colorizedimage` | A colour composite of one or more fluorescence image channels saved as a single-page, single-file colour TIFF or JPEG. _Optional_ |
+| `darkimage` | Dark background fluorescence microscopy image. _Optional_ |
+
+Here is a samplesheet example for one sample:
```csv title="samplesheet.csv"
-sample,fastq_1,fastq_2
-CONTROL_REP1,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz
-CONTROL_REP2,AEG588A2_S2_L002_R1_001.fastq.gz,AEG588A2_S2_L002_R2_001.fastq.gz
-CONTROL_REP3,AEG588A3_S3_L002_R1_001.fastq.gz,AEG588A3_S3_L002_R2_001.fastq.gz
-TREATMENT_REP1,AEG588A4_S4_L003_R1_001.fastq.gz,
-TREATMENT_REP2,AEG588A5_S5_L003_R1_001.fastq.gz,
-TREATMENT_REP3,AEG588A6_S6_L003_R1_001.fastq.gz,
-TREATMENT_REP3,AEG588A6_S6_L004_R1_001.fastq.gz,
+sample,fastq_dir,image,cytaimage,slide,area
+Visium_HD_Human_Lung_Cancer_Fixed_Frozen,Visium_HD_Human_Lung_Cancer_Fixed_Frozen_fastqs,Visium_HD_Human_Lung_Cancer_Fixed_Frozen_tissue_image.btf,Visium_HD_Human_Lung_Cancer_Fixed_Frozen_image.tif,H1-TY834G7,D1
```
-| Column | Description |
-| --------- | -------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
-| `sample` | Custom sample name. This entry will be identical for multiple sequencing libraries/runs from the same sample. Spaces in sample names are automatically converted to underscores (`_`). |
-| `fastq_1` | Full path to FastQ file for Illumina short reads 1. File has to be gzipped and have the extension ".fastq.gz" or ".fq.gz". |
-| `fastq_2` | Full path to FastQ file for Illumina short reads 2. File has to be gzipped and have the extension ".fastq.gz" or ".fq.gz". |
+This samplesheet was made for [this public sample](https://www.10xgenomics.com/datasets/visium-hd-cytassist-gene-expression-human-lung-cancer-fixed-frozen) (download all the "Input files" and untar the `fastq` zip file to test it).
-An [example samplesheet](../assets/samplesheet.csv) has been provided with the pipeline.
+## Sopa parameters
+
+You'll also need to choose some Sopa parameters that you'll provide to Nextflow via the `-params-file` option. You can find existing Sopa parameter files [here](https://github.com/gustaveroussy/sopa/tree/main/workflow/config), and follow the [corresponding README instructions](https://github.com/gustaveroussy/sopa/blob/main/workflow/config/README.md) of to get your `-params-file` argument.
+
+For instance, if you have Xenium data and want to run Sopa with `proseg`, you can use:
+
+```
+-params-file https://raw.githubusercontent.com/gustaveroussy/sopa/refs/heads/main/workflow/config/xenium/proseg.yaml
+```
+
+> [!NOTE]
+> This `-params-file` option is **not** specific to Sopa - you can list other Nextflow params inside it. In that case, make your own local params-file.
## Running the pipeline
-The typical command for running the pipeline is as follows:
+Once you have defined your samplesheet and `params-file`, you'll be able to run `nf-core/sopa`. The typical command for running the pipeline is as follows:
```bash
-nextflow run nf-core/sopa --input ./samplesheet.csv --outdir ./results -profile docker
+nextflow run nf-core/sopa --input ./samplesheet.csv -params-file --outdir ./results -profile docker
```
+> [!NOTE]
+> For Visium HD data, you may also need to provide a `--spaceranger_probeset` argument with an official 10X Genomics probe set (see [here](https://www.10xgenomics.com/support/software/space-ranger/downloads)). For instance, you can use:
+>
+> ```
+> --spaceranger_probeset https://cf.10xgenomics.com/supp/spatial-exp/probeset/Visium_Human_Transcriptome_Probe_Set_v2.0_GRCh38-2020-A.csv
+> ```
+
This will launch the pipeline with the `docker` configuration profile. See below for more information about profiles.
Note that the pipeline will create the following files in your working directory:
diff --git a/main.nf b/main.nf
index 0cbc95d..eec0b15 100644
--- a/main.nf
+++ b/main.nf
@@ -15,32 +15,9 @@
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
*/
-include { SOPA } from './workflows/sopa'
+include { SOPA } from './workflows/sopa'
include { PIPELINE_INITIALISATION } from './subworkflows/local/utils_nfcore_sopa_pipeline'
-include { PIPELINE_COMPLETION } from './subworkflows/local/utils_nfcore_sopa_pipeline'
-/*
-~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
- NAMED WORKFLOWS FOR PIPELINE
-~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
-*/
-
-//
-// WORKFLOW: Run main analysis pipeline depending on type of input
-//
-workflow NFCORE_SOPA {
-
- take:
- samplesheet // channel: samplesheet read in from --input
-
- main:
-
- //
- // WORKFLOW: Run pipeline
- //
- SOPA (
- samplesheet
- )
-}
+include { PIPELINE_COMPLETION } from './subworkflows/local/utils_nfcore_sopa_pipeline'
/*
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
RUN MAIN WORKFLOW
@@ -48,12 +25,10 @@ workflow NFCORE_SOPA {
*/
workflow {
-
- main:
//
// SUBWORKFLOW: Run initialisation tasks
//
- PIPELINE_INITIALISATION (
+ PIPELINE_INITIALISATION(
params.version,
params.validate_params,
params.monochrome_logs,
@@ -68,13 +43,13 @@ workflow {
//
// WORKFLOW: Run main workflow
//
- NFCORE_SOPA (
+ NFCORE_SOPA(
PIPELINE_INITIALISATION.out.samplesheet
)
//
// SUBWORKFLOW: Run completion tasks
//
- PIPELINE_COMPLETION (
+ PIPELINE_COMPLETION(
params.email,
params.email_on_fail,
params.plaintext_email,
@@ -83,9 +58,25 @@ workflow {
params.hook_url,
)
}
-
/*
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
- THE END
+ NAMED WORKFLOWS FOR PIPELINE
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
*/
+
+//
+// WORKFLOW: Run main analysis pipeline depending on type of input
+//
+workflow NFCORE_SOPA {
+ take:
+ samplesheet // channel: samplesheet read in from --input
+
+ main:
+
+ //
+ // WORKFLOW: Run pipeline
+ //
+ SOPA(
+ samplesheet
+ )
+}
diff --git a/modules.json b/modules.json
index fbad5e6..8054cad 100644
--- a/modules.json
+++ b/modules.json
@@ -4,7 +4,18 @@
"repos": {
"https://github.com/nf-core/modules.git": {
"modules": {
- "nf-core": {}
+ "nf-core": {
+ "spaceranger/count": {
+ "branch": "master",
+ "git_sha": "41dfa3f7c0ffabb96a6a813fe321c6d1cc5b6e46",
+ "installed_by": ["modules"]
+ },
+ "untar": {
+ "branch": "master",
+ "git_sha": "41dfa3f7c0ffabb96a6a813fe321c6d1cc5b6e46",
+ "installed_by": ["modules"]
+ }
+ }
},
"subworkflows": {
"nf-core": {
diff --git a/modules/local/aggregate/environment.yml b/modules/local/aggregate/environment.yml
new file mode 100644
index 0000000..0f643ba
--- /dev/null
+++ b/modules/local/aggregate/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
diff --git a/modules/local/aggregate/main.nf b/modules/local/aggregate/main.nf
new file mode 100644
index 0000000..d5ec577
--- /dev/null
+++ b/modules/local/aggregate/main.nf
@@ -0,0 +1,21 @@
+process AGGREGATE {
+ label "process_medium"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8'
+ : 'docker.io/quentinblampey/sopa:2.1.8'}"
+
+ input:
+ tuple val(meta), path(sdata_path)
+ val cli_arguments
+
+ output:
+ tuple val(meta), path(sdata_path)
+ path "${sdata_path}/tables/table"
+
+ script:
+ """
+ sopa aggregate ${sdata_path} ${cli_arguments}
+ """
+}
diff --git a/modules/local/explorer/environment.yml b/modules/local/explorer/environment.yml
new file mode 100644
index 0000000..0f643ba
--- /dev/null
+++ b/modules/local/explorer/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
diff --git a/modules/local/explorer/main.nf b/modules/local/explorer/main.nf
new file mode 100644
index 0000000..053c339
--- /dev/null
+++ b/modules/local/explorer/main.nf
@@ -0,0 +1,26 @@
+process EXPLORER {
+ label "process_high"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8'
+ : 'docker.io/quentinblampey/sopa:2.1.8'}"
+
+ publishDir "${params.outdir}", mode: params.publish_dir_mode
+
+ input:
+ tuple val(meta), path(sdata_path)
+ val cli_arguments
+
+ output:
+ path "${meta.explorer_dir}/experiment.xenium"
+ path "${meta.explorer_dir}/analysis.zarr.zip"
+ path "${meta.explorer_dir}/cell_feature_matrix.zarr.zip"
+ path "${meta.explorer_dir}/adata.h5ad"
+ path "${meta.explorer_dir}/cells.zarr.zip"
+
+ script:
+ """
+ sopa explorer write ${sdata_path} --output-path ${meta.explorer_dir} ${cli_arguments} --mode "-it"
+ """
+}
diff --git a/modules/local/explorer_raw/environment.yml b/modules/local/explorer_raw/environment.yml
new file mode 100644
index 0000000..0f643ba
--- /dev/null
+++ b/modules/local/explorer_raw/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
diff --git a/modules/local/explorer_raw/main.nf b/modules/local/explorer_raw/main.nf
new file mode 100644
index 0000000..c20bcf4
--- /dev/null
+++ b/modules/local/explorer_raw/main.nf
@@ -0,0 +1,24 @@
+include { ArgsExplorerRaw } from '../utils'
+
+process EXPLORER_RAW {
+ label "process_high"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8'
+ : 'docker.io/quentinblampey/sopa:2.1.8'}"
+
+ publishDir "${params.outdir}", mode: params.publish_dir_mode
+
+ input:
+ tuple val(meta), path(sdata_path), path(data_dir)
+
+ output:
+ path "${meta.explorer_dir}/morphology*"
+ path "${meta.explorer_dir}/transcripts*", optional: true
+
+ script:
+ """
+ sopa explorer write ${sdata_path} --output-path ${meta.explorer_dir} ${ArgsExplorerRaw(params, data_dir.toString())} --mode "+it" --no-save-h5ad
+ """
+}
diff --git a/modules/local/fluo_annotation/environment.yml b/modules/local/fluo_annotation/environment.yml
new file mode 100644
index 0000000..0f643ba
--- /dev/null
+++ b/modules/local/fluo_annotation/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
diff --git a/modules/local/fluo_annotation/main.nf b/modules/local/fluo_annotation/main.nf
new file mode 100644
index 0000000..39d5d0a
--- /dev/null
+++ b/modules/local/fluo_annotation/main.nf
@@ -0,0 +1,27 @@
+process FLUO_ANNOTATION {
+ label "process_medium"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8'
+ : 'docker.io/quentinblampey/sopa:2.1.8'}"
+
+ input:
+ tuple val(meta), path(sdata_path)
+ val cli_arguments
+
+ output:
+ tuple val(meta), path(sdata_path)
+ path "${sdata_path}/tables/table/obs"
+ path "versions.yml"
+
+ script:
+ """
+ sopa annotate fluorescence ${sdata_path} ${cli_arguments}
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ sopa: \$(sopa --version)
+ END_VERSIONS
+ """
+}
diff --git a/modules/local/make_image_patches/environment.yml b/modules/local/make_image_patches/environment.yml
new file mode 100644
index 0000000..0f643ba
--- /dev/null
+++ b/modules/local/make_image_patches/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
diff --git a/modules/local/make_image_patches/main.nf b/modules/local/make_image_patches/main.nf
new file mode 100644
index 0000000..27d90e4
--- /dev/null
+++ b/modules/local/make_image_patches/main.nf
@@ -0,0 +1,21 @@
+process MAKE_IMAGE_PATCHES {
+ label "process_single"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8'
+ : 'docker.io/quentinblampey/sopa:2.1.8'}"
+
+ input:
+ tuple val(meta), path(sdata_path)
+ val cli_arguments
+
+ output:
+ tuple val(meta), path(sdata_path), path("${sdata_path}/.sopa_cache/patches_file_image")
+ path "${sdata_path}/shapes/image_patches"
+
+ script:
+ """
+ sopa patchify image ${sdata_path} ${cli_arguments}
+ """
+}
diff --git a/modules/local/make_transcript_patches/environment.yml b/modules/local/make_transcript_patches/environment.yml
new file mode 100644
index 0000000..0f643ba
--- /dev/null
+++ b/modules/local/make_transcript_patches/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
diff --git a/modules/local/make_transcript_patches/main.nf b/modules/local/make_transcript_patches/main.nf
new file mode 100644
index 0000000..9aed5d3
--- /dev/null
+++ b/modules/local/make_transcript_patches/main.nf
@@ -0,0 +1,20 @@
+process MAKE_TRANSCRIPT_PATCHES {
+ label "process_medium"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8'
+ : 'docker.io/quentinblampey/sopa:2.1.8'}"
+
+ input:
+ tuple val(meta), path(sdata_path)
+ val cli_arguments
+
+ output:
+ tuple val(meta), path(sdata_path), path("${sdata_path}/.sopa_cache/patches_file_transcripts"), path("${sdata_path}/.sopa_cache/transcript_patches")
+
+ script:
+ """
+ sopa patchify transcripts ${sdata_path} ${cli_arguments}
+ """
+}
diff --git a/modules/local/patch_segmentation_baysor/environment.yml b/modules/local/patch_segmentation_baysor/environment.yml
new file mode 100644
index 0000000..3a43072
--- /dev/null
+++ b/modules/local/patch_segmentation_baysor/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa[baysor]==2.1.6
diff --git a/modules/local/patch_segmentation_baysor/main.nf b/modules/local/patch_segmentation_baysor/main.nf
new file mode 100644
index 0000000..b7bb250
--- /dev/null
+++ b/modules/local/patch_segmentation_baysor/main.nf
@@ -0,0 +1,21 @@
+process PATCH_SEGMENTATION_BAYSOR {
+ label "process_long"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8-baysor'
+ : 'docker.io/quentinblampey/sopa:2.1.8-baysor'}"
+
+ input:
+ tuple val(meta), path(sdata_path), val(cli_arguments), val(index), val(n_patches)
+
+ output:
+ tuple val(meta), path(sdata_path), path("${sdata_path}/.sopa_cache/transcript_patches/${index}/segmentation_counts.loom"), val(n_patches)
+
+ script:
+ """
+ export JULIA_NUM_THREADS=${task.cpus} # parallelize within each patch for Baysor >= v0.7
+
+ sopa segmentation baysor ${sdata_path} --patch-index ${index} ${cli_arguments}
+ """
+}
diff --git a/modules/local/patch_segmentation_cellpose/environment.yml b/modules/local/patch_segmentation_cellpose/environment.yml
new file mode 100644
index 0000000..91faa94
--- /dev/null
+++ b/modules/local/patch_segmentation_cellpose/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa[cellpose]==2.1.6
diff --git a/modules/local/patch_segmentation_cellpose/main.nf b/modules/local/patch_segmentation_cellpose/main.nf
new file mode 100644
index 0000000..6cb8f5d
--- /dev/null
+++ b/modules/local/patch_segmentation_cellpose/main.nf
@@ -0,0 +1,22 @@
+process PATCH_SEGMENTATION_CELLPOSE {
+ label "process_single"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8-cellpose'
+ : 'docker.io/quentinblampey/sopa:2.1.8-cellpose'}"
+
+ input:
+ tuple val(meta), path(sdata_path), val(cli_arguments), val(index), val(n_patches)
+
+ output:
+ tuple val(meta), path(sdata_path), path("${sdata_path}/.sopa_cache/cellpose_boundaries/${index}.parquet"), val(n_patches)
+
+ script:
+ """
+ mkdir ./cellpose_cache
+ export CELLPOSE_LOCAL_MODELS_PATH=./cellpose_cache
+
+ sopa segmentation cellpose ${sdata_path} --patch-index ${index} ${cli_arguments}
+ """
+}
diff --git a/modules/local/patch_segmentation_comseg/environment.yml b/modules/local/patch_segmentation_comseg/environment.yml
new file mode 100644
index 0000000..2654b07
--- /dev/null
+++ b/modules/local/patch_segmentation_comseg/environment.yml
@@ -0,0 +1,9 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
+ - comseg==1.8.2
diff --git a/modules/local/patch_segmentation_comseg/main.nf b/modules/local/patch_segmentation_comseg/main.nf
new file mode 100644
index 0000000..54aa867
--- /dev/null
+++ b/modules/local/patch_segmentation_comseg/main.nf
@@ -0,0 +1,19 @@
+process PATCH_SEGMENTATION_COMSEG {
+ label "process_long"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8-comseg'
+ : 'docker.io/quentinblampey/sopa:2.1.8-comseg'}"
+
+ input:
+ tuple val(meta), path(sdata_path), val(cli_arguments), val(index), val(n_patches)
+
+ output:
+ tuple val(meta), path(sdata_path), path("${sdata_path}/.sopa_cache/transcript_patches/${index}/segmentation_counts.h5ad"), path("${sdata_path}/.sopa_cache/transcript_patches/${index}/segmentation_polygons.json"), val(n_patches)
+
+ script:
+ """
+ sopa segmentation comseg ${sdata_path} --patch-index ${index} ${cli_arguments}
+ """
+}
diff --git a/modules/local/patch_segmentation_proseg/environment.yml b/modules/local/patch_segmentation_proseg/environment.yml
new file mode 100644
index 0000000..0f643ba
--- /dev/null
+++ b/modules/local/patch_segmentation_proseg/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
diff --git a/modules/local/patch_segmentation_proseg/main.nf b/modules/local/patch_segmentation_proseg/main.nf
new file mode 100644
index 0000000..7eb6c51
--- /dev/null
+++ b/modules/local/patch_segmentation_proseg/main.nf
@@ -0,0 +1,28 @@
+process PATCH_SEGMENTATION_PROSEG {
+ label "process_high"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8-proseg'
+ : 'docker.io/quentinblampey/sopa:2.1.8-proseg'}"
+
+ input:
+ tuple val(meta), path(sdata_path), path(patches_file_transcripts), path(transcript_patches)
+ val cli_arguments
+
+ output:
+ tuple val(meta), path(sdata_path)
+ path "${sdata_path}/shapes/proseg_boundaries"
+ path "versions.yml"
+
+ script:
+ """
+ sopa segmentation proseg ${sdata_path} ${cli_arguments}
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ sopa: \$(sopa --version)
+ proseg: \$(proseg --version | cut -d' ' -f2)
+ END_VERSIONS
+ """
+}
diff --git a/modules/local/patch_segmentation_stardist/environment.yml b/modules/local/patch_segmentation_stardist/environment.yml
new file mode 100644
index 0000000..71db8a5
--- /dev/null
+++ b/modules/local/patch_segmentation_stardist/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa[stardist]==2.1.6
diff --git a/modules/local/patch_segmentation_stardist/main.nf b/modules/local/patch_segmentation_stardist/main.nf
new file mode 100644
index 0000000..755c97e
--- /dev/null
+++ b/modules/local/patch_segmentation_stardist/main.nf
@@ -0,0 +1,19 @@
+process PATCH_SEGMENTATION_STARDIST {
+ label "process_low"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8-stardist'
+ : 'docker.io/quentinblampey/sopa:2.1.8-stardist'}"
+
+ input:
+ tuple val(meta), path(sdata_path), val(cli_arguments), val(index), val(n_patches)
+
+ output:
+ tuple val(meta), path(sdata_path), path("${sdata_path}/.sopa_cache/stardist_boundaries/${index}.parquet"), val(n_patches)
+
+ script:
+ """
+ sopa segmentation stardist ${sdata_path} --patch-index ${index} ${cli_arguments}
+ """
+}
diff --git a/modules/local/report/environment.yml b/modules/local/report/environment.yml
new file mode 100644
index 0000000..0f643ba
--- /dev/null
+++ b/modules/local/report/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
diff --git a/modules/local/report/main.nf b/modules/local/report/main.nf
new file mode 100644
index 0000000..36770e2
--- /dev/null
+++ b/modules/local/report/main.nf
@@ -0,0 +1,26 @@
+process REPORT {
+ label "process_medium"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8'
+ : 'docker.io/quentinblampey/sopa:2.1.8'}"
+
+ publishDir "${params.outdir}", mode: params.publish_dir_mode
+
+ input:
+ tuple val(meta), path(sdata_path)
+
+ output:
+ path sdata_path
+ path "${meta.explorer_dir}/analysis_summary.html"
+
+ script:
+ """
+ mkdir -p ${meta.explorer_dir}
+
+ sopa report ${sdata_path} ${meta.explorer_dir}/analysis_summary.html
+
+ rm -r ${sdata_path}/.sopa_cache || true # clean up cache if existing
+ """
+}
diff --git a/modules/local/resolve_baysor/environment.yml b/modules/local/resolve_baysor/environment.yml
new file mode 100644
index 0000000..3a43072
--- /dev/null
+++ b/modules/local/resolve_baysor/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa[baysor]==2.1.6
diff --git a/modules/local/resolve_baysor/main.nf b/modules/local/resolve_baysor/main.nf
new file mode 100644
index 0000000..9e24a89
--- /dev/null
+++ b/modules/local/resolve_baysor/main.nf
@@ -0,0 +1,30 @@
+process RESOLVE_BAYSOR {
+ label "process_low"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8-baysor'
+ : 'docker.io/quentinblampey/sopa:2.1.8-baysor'}"
+
+ input:
+ tuple val(meta), path(sdata_path)
+ val cli_arguments
+
+ output:
+ tuple val(meta), path(sdata_path)
+ path "${sdata_path}/shapes/baysor_boundaries"
+ path "versions.yml"
+
+ script:
+ """
+ sopa resolve baysor ${sdata_path} ${cli_arguments}
+
+ rm -r ${sdata_path}/.sopa_cache/transcript_patches || true # cleanup large baysor files
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ sopa: \$(sopa --version)
+ baysor: \$(baysor --version)
+ END_VERSIONS
+ """
+}
diff --git a/modules/local/resolve_cellpose/environment.yml b/modules/local/resolve_cellpose/environment.yml
new file mode 100644
index 0000000..91faa94
--- /dev/null
+++ b/modules/local/resolve_cellpose/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa[cellpose]==2.1.6
diff --git a/modules/local/resolve_cellpose/main.nf b/modules/local/resolve_cellpose/main.nf
new file mode 100644
index 0000000..a14ff57
--- /dev/null
+++ b/modules/local/resolve_cellpose/main.nf
@@ -0,0 +1,27 @@
+process RESOLVE_CELLPOSE {
+ label "process_low"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8-cellpose'
+ : 'docker.io/quentinblampey/sopa:2.1.8-cellpose'}"
+
+ input:
+ tuple val(meta), path(sdata_path)
+
+ output:
+ tuple val(meta), path(sdata_path)
+ path "${sdata_path}/shapes/cellpose_boundaries"
+ path "versions.yml"
+
+ script:
+ """
+ sopa resolve cellpose ${sdata_path}
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ sopa: \$(sopa --version)
+ cellpose: \$(cellpose --version | grep 'cellpose version:' | head -n1 | awk '{print \$3}')
+ END_VERSIONS
+ """
+}
diff --git a/modules/local/resolve_comseg/environment.yml b/modules/local/resolve_comseg/environment.yml
new file mode 100644
index 0000000..2654b07
--- /dev/null
+++ b/modules/local/resolve_comseg/environment.yml
@@ -0,0 +1,9 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
+ - comseg==1.8.2
diff --git a/modules/local/resolve_comseg/main.nf b/modules/local/resolve_comseg/main.nf
new file mode 100644
index 0000000..9dcfaef
--- /dev/null
+++ b/modules/local/resolve_comseg/main.nf
@@ -0,0 +1,30 @@
+process RESOLVE_COMSEG {
+ label "process_low"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8-comseg'
+ : 'docker.io/quentinblampey/sopa:2.1.8-comseg'}"
+
+ input:
+ tuple val(meta), path(sdata_path)
+ val cli_arguments
+
+ output:
+ tuple val(meta), path(sdata_path)
+ path "${sdata_path}/shapes/comseg_boundaries"
+ path "versions.yml"
+
+ script:
+ """
+ sopa resolve comseg ${sdata_path} ${cli_arguments}
+
+ rm -r ${sdata_path}/.sopa_cache/transcript_patches || true # cleanup large comseg files
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ sopa: \$(sopa --version)
+ comseg: \$(python -c "import comseg; print(comseg.__version__)" 2> /dev/null)
+ END_VERSIONS
+ """
+}
diff --git a/modules/local/resolve_stardist/environment.yml b/modules/local/resolve_stardist/environment.yml
new file mode 100644
index 0000000..71db8a5
--- /dev/null
+++ b/modules/local/resolve_stardist/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa[stardist]==2.1.6
diff --git a/modules/local/resolve_stardist/main.nf b/modules/local/resolve_stardist/main.nf
new file mode 100644
index 0000000..f0243ed
--- /dev/null
+++ b/modules/local/resolve_stardist/main.nf
@@ -0,0 +1,27 @@
+process RESOLVE_STARDIST {
+ label "process_low"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8-stardist'
+ : 'docker.io/quentinblampey/sopa:2.1.8-stardist'}"
+
+ input:
+ tuple val(meta), path(sdata_path)
+
+ output:
+ tuple val(meta), path(sdata_path)
+ path "${sdata_path}/shapes/stardist_boundaries"
+ path "versions.yml"
+
+ script:
+ """
+ sopa resolve stardist ${sdata_path}
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ sopa: \$(sopa --version)
+ stardist: \$(python -c "import stardist; print(stardist.__version__)" 2> /dev/null)
+ END_VERSIONS
+ """
+}
diff --git a/modules/local/scanpy_preprocess/environment.yml b/modules/local/scanpy_preprocess/environment.yml
new file mode 100644
index 0000000..0f643ba
--- /dev/null
+++ b/modules/local/scanpy_preprocess/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
diff --git a/modules/local/scanpy_preprocess/main.nf b/modules/local/scanpy_preprocess/main.nf
new file mode 100644
index 0000000..a76b604
--- /dev/null
+++ b/modules/local/scanpy_preprocess/main.nf
@@ -0,0 +1,28 @@
+process SCANPY_PREPROCESS {
+ label "process_high"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8'
+ : 'docker.io/quentinblampey/sopa:2.1.8'}"
+
+ input:
+ tuple val(meta), path(sdata_path)
+ val cli_arguments
+
+ output:
+ tuple val(meta), path(sdata_path)
+ path "${sdata_path}/tables/table"
+ path "versions.yml"
+
+ script:
+ """
+ sopa scanpy-preprocess ${sdata_path} ${cli_arguments}
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ sopa: \$(sopa --version)
+ scanpy: \$(python -c "import scanpy; print(scanpy.__version__)" 2> /dev/null)
+ END_VERSIONS
+ """
+}
diff --git a/modules/local/tangram_annotation/environment.yml b/modules/local/tangram_annotation/environment.yml
new file mode 100644
index 0000000..650c28b
--- /dev/null
+++ b/modules/local/tangram_annotation/environment.yml
@@ -0,0 +1,9 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
+ - tangram-sc==1.0.4
diff --git a/modules/local/tangram_annotation/main.nf b/modules/local/tangram_annotation/main.nf
new file mode 100644
index 0000000..a1dae87
--- /dev/null
+++ b/modules/local/tangram_annotation/main.nf
@@ -0,0 +1,29 @@
+process TANGRAM_ANNOTATION {
+ label "process_gpu"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8-tangram'
+ : 'docker.io/quentinblampey/sopa:2.1.8-tangram'}"
+
+ input:
+ tuple val(meta), path(sdata_path)
+ file sc_reference
+ val cli_arguments
+
+ output:
+ tuple val(meta), path(sdata_path)
+ path "${sdata_path}/tables/table/obs"
+ path "versions.yml"
+
+ script:
+ """
+ sopa annotate tangram ${sdata_path} --sc-reference-path ${sc_reference} ${cli_arguments}
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ sopa: \$(sopa --version)
+ tangram: \$(python -c "import tangram; print(tangram.__version__)" 2> /dev/null)
+ END_VERSIONS
+ """
+}
diff --git a/modules/local/tissue_segmentation/environment.yml b/modules/local/tissue_segmentation/environment.yml
new file mode 100644
index 0000000..0f643ba
--- /dev/null
+++ b/modules/local/tissue_segmentation/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
diff --git a/modules/local/tissue_segmentation/main.nf b/modules/local/tissue_segmentation/main.nf
new file mode 100644
index 0000000..38e34a6
--- /dev/null
+++ b/modules/local/tissue_segmentation/main.nf
@@ -0,0 +1,21 @@
+process TISSUE_SEGMENTATION {
+ label "process_low"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8'
+ : 'docker.io/quentinblampey/sopa:2.1.8'}"
+
+ input:
+ tuple val(meta), path(sdata_path)
+ val cli_arguments
+
+ output:
+ tuple val(meta), path(sdata_path)
+ path "${sdata_path}/shapes/region_of_interest"
+
+ script:
+ """
+ sopa segmentation tissue ${sdata_path} ${cli_arguments}
+ """
+}
diff --git a/modules/local/to_spatialdata/environment.yml b/modules/local/to_spatialdata/environment.yml
new file mode 100644
index 0000000..0f643ba
--- /dev/null
+++ b/modules/local/to_spatialdata/environment.yml
@@ -0,0 +1,8 @@
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - python=3.11
+ - pip=24.3.1
+ - pip:
+ - sopa==2.1.6
diff --git a/modules/local/to_spatialdata/main.nf b/modules/local/to_spatialdata/main.nf
new file mode 100644
index 0000000..9de77a1
--- /dev/null
+++ b/modules/local/to_spatialdata/main.nf
@@ -0,0 +1,29 @@
+include { ArgsToSpatialData } from '../utils'
+
+process TO_SPATIALDATA {
+ label "process_high"
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'apptainer' && !task.ext.singularity_pull_docker_container
+ ? 'docker://quentinblampey/sopa:2.1.8'
+ : 'docker.io/quentinblampey/sopa:2.1.8'}"
+
+ input:
+ tuple val(meta), path(data_dir), path(fullres_image_file)
+
+ output:
+ tuple val(meta), path("${meta.sdata_dir}")
+ path "versions.yml"
+
+ script:
+ """
+ sopa convert ${data_dir} --sdata-path ${meta.sdata_dir} ${ArgsToSpatialData(params, meta, fullres_image_file.toString())}
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ sopa: \$(sopa --version)
+ spatialdata: \$(python -c "import spatialdata; print(spatialdata.__version__)" 2> /dev/null)
+ spatialdata_io: \$(python -c "import spatialdata_io; print(spatialdata_io.__version__)" 2> /dev/null)
+ END_VERSIONS
+ """
+}
diff --git a/modules/local/utils.nf b/modules/local/utils.nf
new file mode 100644
index 0000000..6768692
--- /dev/null
+++ b/modules/local/utils.nf
@@ -0,0 +1,89 @@
+def stringifyItem(String key, value) {
+ key = key.replace('_', '-')
+
+ def option = "--${key}"
+
+ if (value instanceof Boolean) {
+ return value ? option : "--no-${key}"
+ }
+ if (value instanceof List) {
+ return value.collect { v -> "${option} ${stringifyValueForCli(v)}" }.join(" ")
+ }
+ if (value instanceof Map) {
+ return "${option} \"" + stringifyValueForCli(value) + "\""
+ }
+ return "${option} ${stringifyValueForCli(value)}"
+}
+
+def stringifyValueForCli(value) {
+ if (value instanceof Map) {
+ return "{" + value.collect { k, v -> "'${k}': ${stringifyValueForCli(v)}" }.join(", ") + "}"
+ }
+ if (value instanceof List) {
+ return "[" + value.collect { stringifyValueForCli(it) }.join(", ") + "]"
+ }
+ if (value instanceof String) {
+ return "'${value}'"
+ }
+ if (value instanceof Boolean) {
+ return value ? "True" : "False"
+ }
+ if (value instanceof Number) {
+ return value.toString()
+ }
+ return "'${value.toString()}'"
+}
+
+def ArgsCLI(Map params, String contains = null, List keys = null) {
+ params = params ?: [:]
+
+ return params
+ .findAll { key, _value ->
+ (contains == null || key.contains(contains)) && (keys == null || key in keys)
+ }
+ .collect { key, value -> stringifyItem(key, value) }
+ .join(" ")
+}
+
+def ArgsToSpatialData(Map params, Map meta, String fullres_image_file) {
+ def args = deepCopyCollection(params.read)
+
+ if (args.technology == "visium_hd") {
+ if (!args.kwargs) {
+ args.kwargs = ["dataset_id": meta.id]
+ }
+ else {
+ args.kwargs["dataset_id"] = meta.id
+ }
+
+ args.kwargs["fullres_image_file"] = fullres_image_file
+ }
+
+ return ArgsCLI(args)
+}
+
+def ArgsExplorerRaw(Map params, String raw_data_path) {
+ def args = deepCopyCollection(params.explorer ?: [:])
+
+ if (params.read.technology == "xenium") {
+ args["raw_data_path"] = raw_data_path
+ }
+
+ return ArgsCLI(args)
+}
+
+def deepCopyCollection(object) {
+ if (object instanceof Map) {
+ object.collectEntries { key, value ->
+ [key, deepCopyCollection(value)]
+ }
+ }
+ else if (object instanceof List) {
+ object.collect { item ->
+ deepCopyCollection(item)
+ }
+ }
+ else {
+ object
+ }
+}
diff --git a/modules/nf-core/spaceranger/count/main.nf b/modules/nf-core/spaceranger/count/main.nf
new file mode 100644
index 0000000..6d882b2
--- /dev/null
+++ b/modules/nf-core/spaceranger/count/main.nf
@@ -0,0 +1,75 @@
+process SPACERANGER_COUNT {
+ tag "$meta.id"
+ label 'process_high'
+
+ container "nf-core/spaceranger:3.1.3"
+
+ input:
+ tuple val(meta), path(reads), path(image), val(slide), val(area), path(cytaimage), path(darkimage), path(colorizedimage), path(alignment), path(slidefile)
+ path(reference)
+ path(probeset)
+
+ output:
+ tuple val(meta), path("outs/**"), emit: outs
+ path "versions.yml", emit: versions
+
+ when:
+ task.ext.when == null || task.ext.when
+
+ script:
+ // Exit if running this module with -profile conda / -profile mamba
+ if (workflow.profile.tokenize(',').intersect(['conda', 'mamba']).size() >= 1) {
+ error "SPACERANGER_COUNT module does not support Conda. Please use Docker / Singularity / Podman instead."
+ }
+ def args = task.ext.args ?: ''
+ def prefix = task.ext.prefix ?: "${meta.id}"
+ // Add flags for optional inputs on demand.
+ def probeset = probeset ? "--probe-set=\"${probeset}\"" : ""
+ def alignment = alignment ? "--loupe-alignment=\"${alignment}\"" : ""
+ def slidefile = slidefile ? "--slidefile=\"${slidefile}\"" : ""
+ def image = image ? "--image=\"${image}\"" : ""
+ def cytaimage = cytaimage ? "--cytaimage=\"${cytaimage}\"" : ""
+ def darkimage = darkimage ? "--darkimage=\"${darkimage}\"" : ""
+ def colorizedimage = colorizedimage ? "--colorizedimage=\"${colorizedimage}\"" : ""
+ if (slide.matches("visium-(.*)") && area == "" && slidefile == "") {
+ slide_and_area = "--unknown-slide=\"${slide}\""
+ } else {
+ slide_and_area = "--slide=\"${slide}\" --area=\"${area}\""
+ }
+ """
+ spaceranger count \\
+ --id="${prefix}" \\
+ --sample="${meta.id}" \\
+ --fastqs=. \\
+ --transcriptome="${reference}" \\
+ --localcores=${task.cpus} \\
+ --localmem=${task.memory.toGiga()} \\
+ $image $cytaimage $darkimage $colorizedimage \\
+ $slide_and_area \\
+ $probeset \\
+ $alignment \\
+ $slidefile \\
+ $args
+ mv ${prefix}/outs outs
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ spaceranger: \$(spaceranger -V | sed -e "s/spaceranger spaceranger-//g")
+ END_VERSIONS
+ """
+
+ stub:
+ // Exit if running this module with -profile conda / -profile mamba
+ if (workflow.profile.tokenize(',').intersect(['conda', 'mamba']).size() >= 1) {
+ error "SPACERANGER_COUNT module does not support Conda. Please use Docker / Singularity / Podman instead."
+ }
+ """
+ mkdir -p outs/
+ touch outs/fake_file.txt
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ spaceranger: \$(spaceranger -V | sed -e "s/spaceranger spaceranger-//g")
+ END_VERSIONS
+ """
+}
diff --git a/modules/nf-core/spaceranger/count/meta.yml b/modules/nf-core/spaceranger/count/meta.yml
new file mode 100644
index 0000000..cf6efb5
--- /dev/null
+++ b/modules/nf-core/spaceranger/count/meta.yml
@@ -0,0 +1,119 @@
+# yaml-language-server: $schema=https://raw.githubusercontent.com/nf-core/modules/master/modules/yaml-schema.json
+name: "spaceranger_count"
+description: Module to use the 10x Space Ranger pipeline to process 10x spatial transcriptomics
+ data
+keywords:
+ - align
+ - count
+ - spatial
+ - spaceranger
+ - imaging
+tools:
+ - "spaceranger":
+ description: |
+ Visium Spatial Gene Expression is a next-generation molecular profiling solution for classifying tissue
+ based on total mRNA. Space Ranger is a set of analysis pipelines that process Visium Spatial Gene Expression
+ data with brightfield and fluorescence microscope images. Space Ranger allows users to map the whole
+ transcriptome in formalin fixed paraffin embedded (FFPE) and fresh frozen tissues to discover novel
+ insights into normal development, disease pathology, and clinical translational research. Space Ranger provides
+ pipelines for end to end analysis of Visium Spatial Gene Expression experiments.
+ homepage: "https://support.10xgenomics.com/spatial-gene-expression/software/pipelines/latest/what-is-space-ranger"
+ documentation: "https://support.10xgenomics.com/spatial-gene-expression/software/pipelines/latest/what-is-space-ranger"
+ tool_dev_url: "https://support.10xgenomics.com/spatial-gene-expression/software/pipelines/latest/what-is-space-ranger"
+ licence:
+ - "10x Genomics EULA"
+ identifier: ""
+input:
+ - - meta:
+ type: map
+ description: |
+ Groovy Map containing sample information
+ e.g. [ id:'test', slide:'10L13-020', area: 'B1']
+
+ `id`, `slide` and `area` are mandatory information!
+ - reads:
+ type: file
+ description: |
+ List of input FastQ files of size 1 and 2 for single-end and paired-end data,
+ respectively.
+ pattern: "${Sample_Name}_S1_L00${Lane_Number}_${I1,I2,R1,R2}_001.fastq.gz"
+ ontologies: []
+ - image:
+ type: file
+ description: Brightfield tissue H&E image in JPEG or TIFF format.
+ pattern: "*.{tif,tiff,jpg,jpeg}"
+ ontologies: []
+ - slide:
+ type: string
+ description: Visium slide ID used for the sample.
+ - area:
+ type: string
+ description: Visium slide capture area used for the sample.
+ - cytaimage:
+ type: file
+ description: |
+ CytAssist instrument captured eosin stained Brightfield tissue image with fiducial
+ frame in TIFF format. The size of this image is set at 3k in both dimensions and this image should
+ not be modified any way before passing it as input to either Space Ranger or Loupe Browser.
+ pattern: "*.{tif,tiff}"
+ ontologies: []
+ - darkimage:
+ type: file
+ description: |
+ Optional for dark background fluorescence microscope image input. Multi-channel, dark-background fluorescence
+ image as either a single, multi-layer TIFF file or as multiple TIFF or JPEG files.
+ pattern: "*.{tif,tiff,jpg,jpeg}"
+ ontologies: []
+ - colorizedimage:
+ type: file
+ description: |
+ Required for color composite fluorescence microscope image input.
+ A color composite of one or more fluorescence image channels saved as a single-page,
+ single-file color TIFF or JPEG.
+ pattern: "*.{tif,tiff,jpg,jpeg}"
+ ontologies: []
+ - alignment:
+ type: file
+ description: OPTIONAL - Path to manual image alignment.
+ pattern: "*.json"
+ ontologies:
+ - edam: http://edamontology.org/format_3464 # JSON
+ - slidefile:
+ type: file
+ description: OPTIONAL - Path to slide specifications.
+ pattern: "*.json"
+ ontologies:
+ - edam: http://edamontology.org/format_3464 # JSON
+ - reference:
+ type: directory
+ description: Folder containing all the reference indices needed by Space Ranger
+ - probeset:
+ type: file
+ description: OPTIONAL - Probe set specification.
+ pattern: "*.csv"
+ ontologies:
+ - edam: http://edamontology.org/format_3752 # CSV
+output:
+ outs:
+ - - meta:
+ type: map
+ description: |
+ Groovy Map containing sample information
+ e.g. [ id:'test', single_end:false ]
+ - outs/**:
+ type: file
+ description: Files containing the outputs of Space Ranger, see official 10X
+ Genomics documentation for a complete list
+ pattern: "outs/*"
+ ontologies: []
+ versions:
+ - versions.yml:
+ type: file
+ description: File containing software versions
+ pattern: "versions.yml"
+ ontologies:
+ - edam: http://edamontology.org/format_3750 # YAML
+authors:
+ - "@grst"
+maintainers:
+ - "@grst"
diff --git a/modules/nf-core/spaceranger/count/tests/main.nf.test b/modules/nf-core/spaceranger/count/tests/main.nf.test
new file mode 100644
index 0000000..3d65e68
--- /dev/null
+++ b/modules/nf-core/spaceranger/count/tests/main.nf.test
@@ -0,0 +1,228 @@
+nextflow_process {
+
+ name "Test Process SPACERANGER_COUNT"
+ script "../main.nf"
+ config "./nextflow.config"
+ process "SPACERANGER_COUNT"
+
+ tag "modules"
+ tag "modules_nfcore"
+ tag "spaceranger"
+ tag "spaceranger/count"
+ tag "spaceranger/mkgtf"
+ tag "spaceranger/mkref"
+
+ test("spaceranger v1 - homo_sapiens - fasta - gtf - fastq - tif - csv") {
+
+ setup {
+ run("SPACERANGER_MKGTF") {
+ script "../../mkgtf/main.nf"
+ process {
+ """
+ input[0] = file(params.modules_testdata_base_path + 'genomics/homo_sapiens/genome/genome.gtf', checkIfExists: true)
+ """
+ }
+ }
+ }
+
+ setup {
+ run("SPACERANGER_MKREF") {
+ script "../../mkref/main.nf"
+ process {
+ """
+ input[0] = file(params.modules_testdata_base_path + 'genomics/homo_sapiens/genome/genome.fasta', checkIfExists: true)
+ input[1] = file(params.modules_testdata_base_path + 'genomics/homo_sapiens/genome/genome.gtf', checkIfExists: true)
+ input[2] = 'homo_sapiens_chr22_reference'
+ """
+ }
+ }
+ }
+
+ when {
+ process {
+ """
+ input[0] = [
+ [
+ id: 'Visium_FFPE_Human_Ovarian_Cancer'
+ ], // Meta map
+ [
+ file(params.modules_testdata_base_path + 'genomics/homo_sapiens/10xgenomics/spaceranger/human-ovarian-cancer-1-standard_v1_ffpe/Visium_FFPE_Human_Ovarian_Cancer_S1_L001_R1_001.fastq.gz', checkIfExists: true),
+ file(params.modules_testdata_base_path + 'genomics/homo_sapiens/10xgenomics/spaceranger/human-ovarian-cancer-1-standard_v1_ffpe/Visium_FFPE_Human_Ovarian_Cancer_S1_L001_R2_001.fastq.gz', checkIfExists: true)
+ ], // Reads
+ file(params.modules_testdata_base_path + 'genomics/homo_sapiens/10xgenomics/spaceranger/human-ovarian-cancer-1-standard_v1_ffpe/Visium_FFPE_Human_Ovarian_Cancer_image.jpg', checkIfExists: true), // Image
+ 'V10L13-020', // Slide
+ 'D1', // Area
+ [], // Cytaimage
+ [], // Darkimage
+ [], // Colorizedimage
+ [], // Manual alignment (default: automatic alignment)
+ [], // Slide specification (default: automatic download)
+ ]
+ input[1] = SPACERANGER_MKREF.out.reference // Reference
+ input[2] = [] // Probeset (default: use the one included with Space Ranger)
+ """
+ }
+ }
+
+ then {
+ assertAll(
+ { assert process.success },
+ { assert snapshot(
+ process.out.versions,
+ process.out.outs.get(0).get(1).findAll { file(it).name !in [
+ 'web_summary.html',
+ 'scalefactors_json.json',
+ 'barcodes.tsv.gz',
+ 'features.tsv.gz',
+ 'matrix.mtx.gz'
+ ]}
+ ).match()
+ },
+ { assert file(process.out.outs.get(0).get(1).find { file(it).name == 'web_summary.html' }).exists() },
+ { assert file(process.out.outs.get(0).get(1).find { file(it).name == 'scalefactors_json.json' }).exists() },
+ { assert file(process.out.outs.get(0).get(1).find { file(it).name == 'barcodes.tsv.gz' }).exists() },
+ { assert file(process.out.outs.get(0).get(1).find { file(it).name == 'features.tsv.gz' }).exists() },
+ { assert file(process.out.outs.get(0).get(1).find { file(it).name == 'matrix.mtx.gz' }).exists() }
+ )
+ }
+ }
+
+ test("spaceranger v1 (stub) - homo_sapiens - fasta - gtf - fastq - tif - csv") {
+
+ setup {
+ run("SPACERANGER_MKGTF") {
+ script "../../mkgtf/main.nf"
+ process {
+ """
+ input[0] = file(params.modules_testdata_base_path + 'genomics/homo_sapiens/genome/genome.gtf', checkIfExists: true)
+ """
+ }
+ }
+ }
+
+ setup {
+ run("SPACERANGER_MKREF") {
+ script "../../mkref/main.nf"
+ process {
+ """
+ input[0] = file(params.modules_testdata_base_path + 'genomics/homo_sapiens/genome/genome.fasta', checkIfExists: true)
+ input[1] = file(params.modules_testdata_base_path + 'genomics/homo_sapiens/genome/genome.gtf', checkIfExists: true)
+ input[2] = 'homo_sapiens_chr22_reference'
+ """
+ }
+ }
+ }
+
+ options "-stub"
+
+ when {
+ process {
+ """
+ input[0] = [
+ [
+ id: 'Visium_FFPE_Human_Ovarian_Cancer'
+ ], // Meta map
+ [
+ file(params.modules_testdata_base_path + 'genomics/homo_sapiens/10xgenomics/spaceranger/human-ovarian-cancer-1-standard_v1_ffpe/Visium_FFPE_Human_Ovarian_Cancer_S1_L001_R1_001.fastq.gz', checkIfExists: true),
+ file(params.modules_testdata_base_path + 'genomics/homo_sapiens/10xgenomics/spaceranger/human-ovarian-cancer-1-standard_v1_ffpe/Visium_FFPE_Human_Ovarian_Cancer_S1_L001_R2_001.fastq.gz', checkIfExists: true)
+ ], // Reads
+ file(params.modules_testdata_base_path + 'genomics/homo_sapiens/10xgenomics/spaceranger/human-ovarian-cancer-1-standard_v1_ffpe/Visium_FFPE_Human_Ovarian_Cancer_image.jpg', checkIfExists: true), // Image
+ 'V10L13-020', // Slide
+ 'D1', // Area
+ [], // Cytaimage
+ [], // Darkimage
+ [], // Colorizedimage
+ [], // Manual alignment (default: automatic alignment)
+ [], // Slide specification (default: automatic download)
+ ]
+ input[1] = SPACERANGER_MKREF.out.reference // Reference
+ input[2] = [] // Probeset (default: use the one included with Space Ranger)
+ """
+ }
+ }
+
+ then {
+ assertAll(
+ { assert process.success },
+ { assert snapshot(process.out.versions).match() },
+ )
+ }
+ }
+
+ test("spaceranger v2 - homo_sapiens - fasta - gtf - fastq - tif - csv") {
+ setup {
+ run("SPACERANGER_MKGTF") {
+ script "../../mkgtf/main.nf"
+ process {
+ """
+ input[0] = file(params.modules_testdata_base_path + 'genomics/homo_sapiens/genome/genome.gtf', checkIfExists: true)
+ """
+ }
+ }
+ }
+
+ setup {
+ run("SPACERANGER_MKREF") {
+ script "../../mkref/main.nf"
+ process {
+ """
+ input[0] = file(params.modules_testdata_base_path + 'genomics/homo_sapiens/genome/genome.fasta', checkIfExists: true)
+ input[1] = file(params.modules_testdata_base_path + 'genomics/homo_sapiens/genome/genome.gtf', checkIfExists: true)
+ input[2] = 'homo_sapiens_chr22_reference'
+ """
+ }
+ }
+ }
+
+ when {
+ process {
+ """
+ input[0] = [
+ [
+ id: 'CytAssist_11mm_FFPE_Human_Glioblastoma_2'
+ ], // Meta map
+ [
+ file(params.modules_testdata_base_path + 'genomics/homo_sapiens/10xgenomics/spaceranger/human-brain-cancer-11-mm-capture-area-ffpe-2-standard_v2_ffpe_cytassist/CytAssist_11mm_FFPE_Human_Glioblastoma_2_S1_L001_R1_001.fastq.gz', checkIfExists: true),
+ file(params.modules_testdata_base_path + 'genomics/homo_sapiens/10xgenomics/spaceranger/human-brain-cancer-11-mm-capture-area-ffpe-2-standard_v2_ffpe_cytassist/CytAssist_11mm_FFPE_Human_Glioblastoma_2_S1_L001_R2_001.fastq.gz', checkIfExists: true)
+ ], // Reads
+ [], // Image
+ 'V52Y10-317', // Slide
+ 'B1', // Area
+ file(params.modules_testdata_base_path + 'genomics/homo_sapiens/10xgenomics/spaceranger/human-brain-cancer-11-mm-capture-area-ffpe-2-standard_v2_ffpe_cytassist/CytAssist_11mm_FFPE_Human_Glioblastoma_image.tif', checkIfExists: true), // Cytaimage
+ [], // Darkimage
+ [], // Colorizedimage
+ [], // Manual alignment (default: automatic alignment)
+ file('https://s3.us-west-2.amazonaws.com/10x.spatial-slides/gpr/V52Y10/V52Y10-317.gpr') // Slide specification (default: automatic download)
+ ]
+ input[1] = SPACERANGER_MKREF.out.reference // Reference
+ input[2] = file(params.modules_testdata_base_path + 'genomics/homo_sapiens/10xgenomics/spaceranger/human-brain-cancer-11-mm-capture-area-ffpe-2-standard_v2_ffpe_cytassist/CytAssist_11mm_FFPE_Human_Glioblastoma_probe_set.csv', checkIfExists: true) // Probeset (default: use the one included with Space Ranger)
+ """
+ }
+ }
+
+ then {
+ assertAll(
+ { assert process.success },
+ { assert snapshot(
+ process.out.versions,
+ process.out.outs.get(0).get(1).findAll { file(it).name !in [
+ 'web_summary.html',
+ 'scalefactors_json.json',
+ 'molecule_info.h5',
+ 'barcodes.tsv.gz',
+ 'features.tsv.gz',
+ 'matrix.mtx.gz',
+ 'cloupe.cloupe'
+ ]}
+ ).match()
+ },
+ { assert file(process.out.outs.get(0).get(1).find { file(it).name == 'web_summary.html' }).exists() },
+ { assert file(process.out.outs.get(0).get(1).find { file(it).name == 'scalefactors_json.json' }).exists() },
+ { assert file(process.out.outs.get(0).get(1).find { file(it).name == 'molecule_info.h5' }).exists() },
+ { assert file(process.out.outs.get(0).get(1).find { file(it).name == 'barcodes.tsv.gz' }).exists() },
+ { assert file(process.out.outs.get(0).get(1).find { file(it).name == 'features.tsv.gz' }).exists() },
+ { assert file(process.out.outs.get(0).get(1).find { file(it).name == 'matrix.mtx.gz' }).exists() }
+ )
+ }
+ }
+}
\ No newline at end of file
diff --git a/modules/nf-core/spaceranger/count/tests/main.nf.test.snap b/modules/nf-core/spaceranger/count/tests/main.nf.test.snap
new file mode 100644
index 0000000..dbfaadf
--- /dev/null
+++ b/modules/nf-core/spaceranger/count/tests/main.nf.test.snap
@@ -0,0 +1,90 @@
+{
+ "spaceranger v1 (stub) - homo_sapiens - fasta - gtf - fastq - tif - csv": {
+ "content": [
+ [
+ "versions.yml:md5,4abe169f33d7f99d5d9876b189060aae"
+ ]
+ ],
+ "meta": {
+ "nf-test": "0.9.1",
+ "nextflow": "24.10.4"
+ },
+ "timestamp": "2025-02-13T09:02:47.710975472"
+ },
+ "spaceranger v2 - homo_sapiens - fasta - gtf - fastq - tif - csv": {
+ "content": [
+ [
+ "versions.yml:md5,4abe169f33d7f99d5d9876b189060aae"
+ ],
+ [
+ "clusters.csv:md5,221a4554e62ea94b0df8dbf637d2c13c",
+ "clusters.csv:md5,53ee76645943b5562392aac51d2d9f3f",
+ "clusters.csv:md5,b791359469683ad19cdb8d1af3de5705",
+ "clusters.csv:md5,9a4f9148e0e834c1127bf8393ece6330",
+ "clusters.csv:md5,c11bcc64f870469ab2f136d9272a7a6d",
+ "clusters.csv:md5,488846bbb469365e199928c7a440320a",
+ "clusters.csv:md5,5941f7e847d35a4f06d3631e21d2eb9d",
+ "clusters.csv:md5,d244d405c32766339d2b7a3fa8bf8cee",
+ "clusters.csv:md5,981386408cd953548994c31253e787de",
+ "clusters.csv:md5,24c4f13449e5362fcbcd41b9ff413992",
+ "differential_expression.csv:md5,589c1bd4529f092bb1d332e7da561dad",
+ "differential_expression.csv:md5,d9d978b398b33ac9687b44531909e0cd",
+ "differential_expression.csv:md5,4edbc893280f9d03c3de00a503e86f8c",
+ "differential_expression.csv:md5,316181d501c495384016227309856b09",
+ "differential_expression.csv:md5,dae49941396609fb08df13b82fe89151",
+ "differential_expression.csv:md5,4a13ae44c8454dbcb0298eb63df8b8e8",
+ "differential_expression.csv:md5,eeb02c4afe1f49d5502fb024b25b2c38",
+ "differential_expression.csv:md5,9a456828fe5d762e6e07383da5c2791d",
+ "differential_expression.csv:md5,bcbd1504976824e9f4d20a8dd36e2a1f",
+ "differential_expression.csv:md5,3ad93fc4d52950cfede885dc58cd2823",
+ "components.csv:md5,811a32dce6c795e958dc4bc635ee53be",
+ "dispersion.csv:md5,64c2e57ef0ca9a80cce8b952c81b62f5",
+ "features_selected.csv:md5,bd0c0a20b0b0009df796e8a755d028c1",
+ "projection.csv:md5,e530c925a185965514fa82f4da83fa81",
+ "variance.csv:md5,4159711ab5d64e97737fad9d75d945b3",
+ "projection.csv:md5,ce729f7e237df4570ac3e4a79251df24",
+ "projection.csv:md5,fa7bdefa8424b233fe6461129ab76d57",
+ "filtered_feature_bc_matrix.h5:md5,704256e5150522d9cf2e75e7e47221b6",
+ "metrics_summary.csv:md5,5ece84f5f8e08839749b1c8f2bff6701",
+ "probe_set.csv:md5,5bfb8f12319be1b2b6c14142537c3804",
+ "raw_feature_bc_matrix.h5:md5,ac24486662643ea68562c1a51cbbb2bd",
+ "raw_probe_bc_matrix.h5:md5,8ab08437814506f98e3f10107cfc38ac",
+ "aligned_fiducials.jpg:md5,51dcc3a32d3d5ca4704f664c8ede81ef",
+ "cytassist_image.tiff:md5,0fb04a55e5658f4d158d986a334b034d",
+ "detected_tissue_image.jpg:md5,11c9fa90913b5c6e93cecdb8f53d58db",
+ "spatial_enrichment.csv:md5,4379bc4fef891b45ff9264ee8c408bd0",
+ "tissue_hires_image.png:md5,834706fff299024fab48e6366afc9cb9",
+ "tissue_lowres_image.png:md5,8c1fcb378f7f886301f49ffc4f84360a",
+ "tissue_positions.csv:md5,930aeb2b790032337d91dd27cc70f135"
+ ]
+ ],
+ "meta": {
+ "nf-test": "0.9.1",
+ "nextflow": "24.10.4"
+ },
+ "timestamp": "2025-02-12T11:24:51.984883864"
+ },
+ "spaceranger v1 - homo_sapiens - fasta - gtf - fastq - tif - csv": {
+ "content": [
+ [
+ "versions.yml:md5,4abe169f33d7f99d5d9876b189060aae"
+ ],
+ [
+ "filtered_feature_bc_matrix.h5:md5,649ac955bcb372b0b767013071cca72c",
+ "metrics_summary.csv:md5,38774fc5f54873d711b4898a2dd50e72",
+ "molecule_info.h5:md5,88bb948a426041165b2cc5fe8b180c21",
+ "raw_feature_bc_matrix.h5:md5,63324ae38fbf28bcc2114f170e0fde5d",
+ "aligned_fiducials.jpg:md5,f6217ddd707bb189e665f56b130c3da8",
+ "detected_tissue_image.jpg:md5,c1c7e8741701a576c1ec103c1aaf98ea",
+ "tissue_hires_image.png:md5,d91f8f176ae35ab824ede87117ac0889",
+ "tissue_lowres_image.png:md5,475a04208d193191c84d7a3b5d4eb287",
+ "tissue_positions.csv:md5,7f9cb407b3dd69726a12967b979a5624"
+ ]
+ ],
+ "meta": {
+ "nf-test": "0.9.1",
+ "nextflow": "24.10.4"
+ },
+ "timestamp": "2025-02-12T11:08:30.165324139"
+ }
+}
\ No newline at end of file
diff --git a/modules/nf-core/spaceranger/count/tests/nextflow.config b/modules/nf-core/spaceranger/count/tests/nextflow.config
new file mode 100644
index 0000000..fe9d61a
--- /dev/null
+++ b/modules/nf-core/spaceranger/count/tests/nextflow.config
@@ -0,0 +1,5 @@
+process {
+ withName: SPACERANGER_COUNT {
+ ext.args = '--create-bam false'
+ }
+}
diff --git a/modules/nf-core/untar/environment.yml b/modules/nf-core/untar/environment.yml
new file mode 100644
index 0000000..9b926b1
--- /dev/null
+++ b/modules/nf-core/untar/environment.yml
@@ -0,0 +1,12 @@
+---
+# yaml-language-server: $schema=https://raw.githubusercontent.com/nf-core/modules/master/modules/environment-schema.json
+channels:
+ - conda-forge
+ - bioconda
+dependencies:
+ - conda-forge::coreutils=9.5
+ - conda-forge::grep=3.11
+ - conda-forge::gzip=1.13
+ - conda-forge::lbzip2=2.5
+ - conda-forge::sed=4.8
+ - conda-forge::tar=1.34
diff --git a/modules/nf-core/untar/main.nf b/modules/nf-core/untar/main.nf
new file mode 100644
index 0000000..e712ebe
--- /dev/null
+++ b/modules/nf-core/untar/main.nf
@@ -0,0 +1,84 @@
+process UNTAR {
+ tag "${archive}"
+ label 'process_single'
+
+ conda "${moduleDir}/environment.yml"
+ container "${workflow.containerEngine == 'singularity' && !task.ext.singularity_pull_docker_container
+ ? 'https://community-cr-prod.seqera.io/docker/registry/v2/blobs/sha256/52/52ccce28d2ab928ab862e25aae26314d69c8e38bd41ca9431c67ef05221348aa/data'
+ : 'community.wave.seqera.io/library/coreutils_grep_gzip_lbzip2_pruned:838ba80435a629f8'}"
+
+ input:
+ tuple val(meta), path(archive)
+
+ output:
+ tuple val(meta), path("${prefix}"), emit: untar
+ path "versions.yml", emit: versions
+
+ when:
+ task.ext.when == null || task.ext.when
+
+ script:
+ def args = task.ext.args ?: ''
+ def args2 = task.ext.args2 ?: ''
+ prefix = task.ext.prefix ?: (meta.id ? "${meta.id}" : archive.baseName.toString().replaceFirst(/\.tar$/, ""))
+
+ """
+ mkdir ${prefix}
+
+ ## Ensures --strip-components only applied when top level of tar contents is a directory
+ ## If just files or multiple directories, place all in prefix
+ if [[ \$(tar -taf ${archive} | grep -o -P "^.*?\\/" | uniq | wc -l) -eq 1 ]]; then
+ tar \\
+ -C ${prefix} --strip-components 1 \\
+ -xavf \\
+ ${args} \\
+ ${archive} \\
+ ${args2}
+ else
+ tar \\
+ -C ${prefix} \\
+ -xavf \\
+ ${args} \\
+ ${archive} \\
+ ${args2}
+ fi
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ untar: \$(echo \$(tar --version 2>&1) | sed 's/^.*(GNU tar) //; s/ Copyright.*\$//')
+ END_VERSIONS
+ """
+
+ stub:
+ prefix = task.ext.prefix ?: (meta.id ? "${meta.id}" : archive.toString().replaceFirst(/\.[^\.]+(.gz)?$/, ""))
+ """
+ mkdir ${prefix}
+ ## Dry-run untaring the archive to get the files and place all in prefix
+ if [[ \$(tar -taf ${archive} | grep -o -P "^.*?\\/" | uniq | wc -l) -eq 1 ]]; then
+ for i in `tar -tf ${archive}`;
+ do
+ if [[ \$(echo "\${i}" | grep -E "/\$") == "" ]];
+ then
+ touch \${i}
+ else
+ mkdir -p \${i}
+ fi
+ done
+ else
+ for i in `tar -tf ${archive}`;
+ do
+ if [[ \$(echo "\${i}" | grep -E "/\$") == "" ]];
+ then
+ touch ${prefix}/\${i}
+ else
+ mkdir -p ${prefix}/\${i}
+ fi
+ done
+ fi
+
+ cat <<-END_VERSIONS > versions.yml
+ "${task.process}":
+ untar: \$(echo \$(tar --version 2>&1) | sed 's/^.*(GNU tar) //; s/ Copyright.*\$//')
+ END_VERSIONS
+ """
+}
diff --git a/modules/nf-core/untar/meta.yml b/modules/nf-core/untar/meta.yml
new file mode 100644
index 0000000..1b6bf49
--- /dev/null
+++ b/modules/nf-core/untar/meta.yml
@@ -0,0 +1,57 @@
+name: untar
+description: Extract files.
+keywords:
+ - untar
+ - uncompress
+ - extract
+tools:
+ - untar:
+ description: |
+ Extract tar.gz files.
+ documentation: https://www.gnu.org/software/tar/manual/
+ licence: ["GPL-3.0-or-later"]
+ identifier: ""
+input:
+ - - meta:
+ type: map
+ description: |
+ Groovy Map containing sample information
+ e.g. [ id:'test', single_end:false ]
+ - archive:
+ type: file
+ description: File to be untar
+ pattern: "*.{tar}.{gz}"
+ ontologies:
+ - edam: http://edamontology.org/format_3981 # TAR format
+ - edam: http://edamontology.org/format_3989 # GZIP format
+output:
+ untar:
+ - - meta:
+ type: map
+ description: |
+ Groovy Map containing sample information
+ e.g. [ id:'test', single_end:false ]
+ pattern: "*/"
+ - ${prefix}:
+ type: map
+ description: |
+ Groovy Map containing sample information
+ e.g. [ id:'test', single_end:false ]
+ pattern: "*/"
+ versions:
+ - versions.yml:
+ type: file
+ description: File containing software versions
+ pattern: "versions.yml"
+ ontologies:
+ - edam: http://edamontology.org/format_3750 # YAML
+authors:
+ - "@joseespinosa"
+ - "@drpatelh"
+ - "@matthdsm"
+ - "@jfy133"
+maintainers:
+ - "@joseespinosa"
+ - "@drpatelh"
+ - "@matthdsm"
+ - "@jfy133"
diff --git a/modules/nf-core/untar/tests/main.nf.test b/modules/nf-core/untar/tests/main.nf.test
new file mode 100644
index 0000000..c957517
--- /dev/null
+++ b/modules/nf-core/untar/tests/main.nf.test
@@ -0,0 +1,85 @@
+nextflow_process {
+
+ name "Test Process UNTAR"
+ script "../main.nf"
+ process "UNTAR"
+ tag "modules"
+ tag "modules_nfcore"
+ tag "untar"
+
+ test("test_untar") {
+
+ when {
+ process {
+ """
+ input[0] = [ [], file(params.modules_testdata_base_path + 'genomics/sarscov2/genome/db/kraken2.tar.gz', checkIfExists: true) ]
+ """
+ }
+ }
+
+ then {
+ assertAll (
+ { assert process.success },
+ { assert snapshot(process.out).match() },
+ )
+ }
+ }
+
+ test("test_untar_onlyfiles") {
+
+ when {
+ process {
+ """
+ input[0] = [ [], file(params.modules_testdata_base_path + 'generic/tar/hello.tar.gz', checkIfExists: true) ]
+ """
+ }
+ }
+
+ then {
+ assertAll (
+ { assert process.success },
+ { assert snapshot(process.out).match() },
+ )
+ }
+ }
+
+ test("test_untar - stub") {
+
+ options "-stub"
+
+ when {
+ process {
+ """
+ input[0] = [ [], file(params.modules_testdata_base_path + 'genomics/sarscov2/genome/db/kraken2.tar.gz', checkIfExists: true) ]
+ """
+ }
+ }
+
+ then {
+ assertAll (
+ { assert process.success },
+ { assert snapshot(process.out).match() },
+ )
+ }
+ }
+
+ test("test_untar_onlyfiles - stub") {
+
+ options "-stub"
+
+ when {
+ process {
+ """
+ input[0] = [ [], file(params.modules_testdata_base_path + 'generic/tar/hello.tar.gz', checkIfExists: true) ]
+ """
+ }
+ }
+
+ then {
+ assertAll (
+ { assert process.success },
+ { assert snapshot(process.out).match() },
+ )
+ }
+ }
+}
diff --git a/modules/nf-core/untar/tests/main.nf.test.snap b/modules/nf-core/untar/tests/main.nf.test.snap
new file mode 100644
index 0000000..ceb91b7
--- /dev/null
+++ b/modules/nf-core/untar/tests/main.nf.test.snap
@@ -0,0 +1,158 @@
+{
+ "test_untar_onlyfiles": {
+ "content": [
+ {
+ "0": [
+ [
+ [
+
+ ],
+ [
+ "hello.txt:md5,e59ff97941044f85df5297e1c302d260"
+ ]
+ ]
+ ],
+ "1": [
+ "versions.yml:md5,6063247258c56fd271d076bb04dd7536"
+ ],
+ "untar": [
+ [
+ [
+
+ ],
+ [
+ "hello.txt:md5,e59ff97941044f85df5297e1c302d260"
+ ]
+ ]
+ ],
+ "versions": [
+ "versions.yml:md5,6063247258c56fd271d076bb04dd7536"
+ ]
+ }
+ ],
+ "meta": {
+ "nf-test": "0.8.4",
+ "nextflow": "24.04.3"
+ },
+ "timestamp": "2024-07-10T12:04:28.231047"
+ },
+ "test_untar_onlyfiles - stub": {
+ "content": [
+ {
+ "0": [
+ [
+ [
+
+ ],
+ [
+ "hello.txt:md5,d41d8cd98f00b204e9800998ecf8427e"
+ ]
+ ]
+ ],
+ "1": [
+ "versions.yml:md5,6063247258c56fd271d076bb04dd7536"
+ ],
+ "untar": [
+ [
+ [
+
+ ],
+ [
+ "hello.txt:md5,d41d8cd98f00b204e9800998ecf8427e"
+ ]
+ ]
+ ],
+ "versions": [
+ "versions.yml:md5,6063247258c56fd271d076bb04dd7536"
+ ]
+ }
+ ],
+ "meta": {
+ "nf-test": "0.8.4",
+ "nextflow": "24.04.3"
+ },
+ "timestamp": "2024-07-10T12:04:45.773103"
+ },
+ "test_untar - stub": {
+ "content": [
+ {
+ "0": [
+ [
+ [
+
+ ],
+ [
+ "hash.k2d:md5,d41d8cd98f00b204e9800998ecf8427e",
+ "opts.k2d:md5,d41d8cd98f00b204e9800998ecf8427e",
+ "taxo.k2d:md5,d41d8cd98f00b204e9800998ecf8427e"
+ ]
+ ]
+ ],
+ "1": [
+ "versions.yml:md5,6063247258c56fd271d076bb04dd7536"
+ ],
+ "untar": [
+ [
+ [
+
+ ],
+ [
+ "hash.k2d:md5,d41d8cd98f00b204e9800998ecf8427e",
+ "opts.k2d:md5,d41d8cd98f00b204e9800998ecf8427e",
+ "taxo.k2d:md5,d41d8cd98f00b204e9800998ecf8427e"
+ ]
+ ]
+ ],
+ "versions": [
+ "versions.yml:md5,6063247258c56fd271d076bb04dd7536"
+ ]
+ }
+ ],
+ "meta": {
+ "nf-test": "0.8.4",
+ "nextflow": "24.04.3"
+ },
+ "timestamp": "2024-07-10T12:04:36.777441"
+ },
+ "test_untar": {
+ "content": [
+ {
+ "0": [
+ [
+ [
+
+ ],
+ [
+ "hash.k2d:md5,8b8598468f54a7087c203ad0190555d9",
+ "opts.k2d:md5,a033d00cf6759407010b21700938f543",
+ "taxo.k2d:md5,094d5891cdccf2f1468088855c214b2c"
+ ]
+ ]
+ ],
+ "1": [
+ "versions.yml:md5,6063247258c56fd271d076bb04dd7536"
+ ],
+ "untar": [
+ [
+ [
+
+ ],
+ [
+ "hash.k2d:md5,8b8598468f54a7087c203ad0190555d9",
+ "opts.k2d:md5,a033d00cf6759407010b21700938f543",
+ "taxo.k2d:md5,094d5891cdccf2f1468088855c214b2c"
+ ]
+ ]
+ ],
+ "versions": [
+ "versions.yml:md5,6063247258c56fd271d076bb04dd7536"
+ ]
+ }
+ ],
+ "meta": {
+ "nf-test": "0.8.4",
+ "nextflow": "24.04.3"
+ },
+ "timestamp": "2024-07-10T12:04:19.377674"
+ }
+}
\ No newline at end of file
diff --git a/nextflow.config b/nextflow.config
index a3cd049..8d1e552 100644
--- a/nextflow.config
+++ b/nextflow.config
@@ -9,9 +9,19 @@
// Global default params, used in configs
params {
- // TODO nf-core: Specify your pipeline's command line flags
// Input options
input = null
+ read = null
+ segmentation = null
+ patchify = null
+ aggregate = null
+ annotation = null
+ scanpy_preprocess = null
+ explorer = null
+
+ // Spaceranger options
+ spaceranger_reference = "https://cf.10xgenomics.com/supp/spatial-exp/refdata-gex-GRCh38-2020-A.tar.gz"
+ spaceranger_probeset = null
// Boilerplate options
outdir = null
@@ -154,8 +164,11 @@ profiles {
apptainer.runOptions = '--nv'
singularity.runOptions = '--nv'
}
- test { includeConfig 'conf/test.config' }
- test_full { includeConfig 'conf/test_full.config' }
+ test { includeConfig 'conf/test.config' }
+ test_baysor { includeConfig 'conf/test_baysor.config' }
+ test_comseg { includeConfig 'conf/test_comseg.config' }
+ test_cellpose { includeConfig 'conf/test_cellpose.config' }
+ test_full { includeConfig 'conf/test_full.config' }
}
// Load nf-core custom profiles from different institutions
@@ -164,10 +177,6 @@ profiles {
includeConfig params.custom_config_base && (!System.getenv('NXF_OFFLINE') || !params.custom_config_base.startsWith('http')) ? "${params.custom_config_base}/nfcore_custom.config" : "/dev/null"
-// Load nf-core/sopa custom profiles from different institutions.
-// TODO nf-core: Optionally, you can add a pipeline-specific nf-core config at https://github.com/nf-core/configs
-// includeConfig params.custom_config_base && (!System.getenv('NXF_OFFLINE') || !params.custom_config_base.startsWith('http')) ? "${params.custom_config_base}/pipeline/sopa.config" : "/dev/null"
-
// Set default registry for Apptainer, Docker, Podman, Charliecloud and Singularity independent of -profile
// Will not be used unless Apptainer / Docker / Podman / Charliecloud / Singularity are enabled
// Set to your registry if you have a mirror of containers
@@ -187,7 +196,6 @@ env {
PYTHONNOUSERSITE = 1
R_PROFILE_USER = "/.Rprofile"
R_ENVIRON_USER = "/.Renviron"
- JULIA_DEPOT_PATH = "/usr/local/share/julia"
}
// Set bash options
@@ -223,14 +231,13 @@ dag {
manifest {
name = 'nf-core/sopa'
contributors = [
- // TODO nf-core: Update the field with the details of the contributors to your pipeline. New with Nextflow version 24.10.0
[
name: 'Quentin Blampey',
- affiliation: '',
- email: '',
- github: '',
- contribution: [], // List of contribution types ('author', 'maintainer' or 'contributor')
- orcid: ''
+ affiliation: 'CentraleSupélec, Gustave Roussy',
+ email: 'quentin.blampey@gmail.com',
+ github: 'https://github.com/quentinblampey',
+ contribution: ['author', 'maintainer'], // List of contribution types ('author', 'maintainer' or 'contributor')
+ orcid: '0000-0002-3836-2889'
],
]
homePage = 'https://github.com/nf-core/sopa'
diff --git a/nextflow_schema.json b/nextflow_schema.json
index 2e19199..8ca653b 100644
--- a/nextflow_schema.json
+++ b/nextflow_schema.json
@@ -38,6 +38,217 @@
}
}
},
+ "sopa_config": {
+ "title": "Sopa config (or params file) - we recommend providing an existing `-params-file` as detailed in the usage section",
+ "type": "object",
+ "fa_icon": "fas fa-rocket",
+ "description": "Parameters related to Sopa",
+ "required": ["read", "segmentation"],
+ "properties": {
+ "read": {
+ "type": "object",
+ "required": ["technology"],
+ "properties": {
+ "technology": {
+ "type": "string",
+ "description": "Technology used for the spatial data, e.g., 'xenium', 'merscope', ...",
+ "fa_icon": "fas fa-microscope",
+ "enum": [
+ "xenium",
+ "merscope",
+ "cosmx",
+ "visium_hd",
+ "molecular_cartography",
+ "macsima",
+ "phenocycler",
+ "hyperion",
+ "ome_tif",
+ "toy_dataset"
+ ]
+ },
+ "kwargs": {
+ "type": "object",
+ "additionalProperties": true,
+ "fa_icon": "fas fa-gear"
+ }
+ },
+ "additionalProperties": true,
+ "description": "Dictionary of parameters to read the raw spatial inputs, e.g., technology name.",
+ "fa_icon": "fas fa-gear"
+ },
+ "segmentation": {
+ "type": "object",
+ "properties": {
+ "tissue": {
+ "type": "object",
+ "additionalProperties": true,
+ "description": "Dictionary of tissue segmentation parameters.",
+ "fa_icon": "fas fa-gear"
+ },
+ "baysor": {
+ "type": "object",
+ "properties": {
+ "config": {
+ "type": "object",
+ "properties": {
+ "data": {
+ "type": "object",
+ "description": "Baysor config: data section.",
+ "fa_icon": "fas fa-gear",
+ "additionalProperties": true
+ },
+ "segmentation": {
+ "type": "object",
+ "description": "Baysor config: segmentation section.",
+ "fa_icon": "fas fa-gear",
+ "additionalProperties": true
+ }
+ },
+ "description": "Baysor configuration parameters.",
+ "fa_icon": "fas fa-gear"
+ }
+ },
+ "additionalProperties": true,
+ "description": "Dictionary of Baysor parameters."
+ },
+ "comseg": {
+ "type": "object",
+ "properties": {
+ "config": {
+ "type": "object",
+ "properties": {
+ "dict_scale": {
+ "type": "object",
+ "description": "Comseg dict scale section.",
+ "fa_icon": "fas fa-gear",
+ "additionalProperties": true
+ }
+ },
+ "additionalProperties": true,
+ "description": "Comseg configuration parameters.",
+ "fa_icon": "fas fa-gear"
+ }
+ },
+ "additionalProperties": true,
+ "description": "Dictionary of Comseg parameters."
+ },
+ "cellpose": {
+ "type": "object",
+ "additionalProperties": true,
+ "description": "Dictionary of Cellpose parameters.",
+ "fa_icon": "fas fa-gear"
+ },
+ "stardist": {
+ "type": "object",
+ "additionalProperties": true,
+ "description": "Dictionary of Stardist parameters.",
+ "fa_icon": "fas fa-gear"
+ },
+ "proseg": {
+ "type": "object",
+ "additionalProperties": true,
+ "description": "Dictionary of Proseg parameters.",
+ "fa_icon": "fas fa-gear"
+ }
+ },
+ "additionalProperties": true,
+ "description": "Dictionary of segmentation parameters.",
+ "fa_icon": "fas fa-gear"
+ },
+ "patchify": {
+ "type": "object",
+ "additionalProperties": true,
+ "description": "Dictionary of patches parameters.",
+ "fa_icon": "fas fa-gear"
+ },
+ "aggregate": {
+ "type": "object",
+ "additionalProperties": true,
+ "description": "Dictionary of aggregation parameters.",
+ "fa_icon": "fas fa-gear"
+ },
+ "annotation": {
+ "type": "object",
+ "required": ["method", "args"],
+ "properties": {
+ "method": {
+ "type": "string",
+ "description": "Cell type annotation method, either 'tangram' or 'fluorescence'",
+ "enum": ["tangram", "fluorescence"],
+ "fa_icon": "fas fa-gear"
+ },
+ "args": {
+ "type": "object",
+ "description": "Cell type annotation arguments",
+ "fa_icon": "fas fa-gear",
+ "properties": {
+ "marker_cell_dict": {
+ "type": "object",
+ "description": "Dictionary mapping cell type markers to their descriptions",
+ "additionalProperties": {
+ "type": "string"
+ }
+ }
+ },
+ "additionalProperties": true
+ }
+ },
+ "additionalProperties": true,
+ "description": "Dictionary of annotation parameters.",
+ "fa_icon": "fas fa-gear"
+ },
+ "scanpy_preprocess": {
+ "type": "object",
+ "properties": {
+ "resolution": {
+ "type": "number",
+ "description": "Resolution parameter for the Leiden clustering algorithm.",
+ "fa_icon": "fas fa-sliders-h"
+ },
+ "check_counts": {
+ "type": "boolean",
+ "description": "Whether to check counts in Scanpy preprocessing.",
+ "fa_icon": "fas fa-check"
+ }
+ },
+ "additionalProperties": true,
+ "description": "Dictionary of Scanpy preprocessing parameters.",
+ "fa_icon": "fas fa-gear"
+ },
+ "explorer": {
+ "type": "object",
+ "additionalProperties": true,
+ "description": "Dictionary of Xenium Explorer parameters.",
+ "fa_icon": "fas fa-gear"
+ }
+ }
+ },
+ "spaceranger_options": {
+ "title": "Space Ranger options",
+ "type": "object",
+ "fa_icon": "fas fa-rocket",
+ "description": "Options related to Space Ranger execution and raw spatial data processing",
+ "properties": {
+ "spaceranger_probeset": {
+ "type": "string",
+ "format": "file-path",
+ "mimetype": "text/csv",
+ "pattern": "^\\S+\\.csv$",
+ "description": "Location of Space Ranger probeset file.",
+ "fa_icon": "fas fa-file-csv",
+ "exists": true
+ },
+ "spaceranger_reference": {
+ "type": "string",
+ "format": "path",
+ "description": "Location of Space Ranger reference directory. May be packed as `tar.gz` file.",
+ "help_text": "Please see the [10x website](https://support.10xgenomics.com/spatial-gene-expression/software/downloads/latest) to download either of the supported human or mouse references. If not specified the GRCh38 human reference is automatically downladed and used.",
+ "fa_icon": "fas fa-folder-open",
+ "default": "https://cf.10xgenomics.com/supp/spatial-exp/refdata-gex-GRCh38-2020-A.tar.gz",
+ "exists": true
+ }
+ }
+ },
"institutional_config_options": {
"title": "Institutional config options",
"type": "object",
@@ -174,6 +385,12 @@
{
"$ref": "#/$defs/input_output_options"
},
+ {
+ "$ref": "#/$defs/sopa_config"
+ },
+ {
+ "$ref": "#/$defs/spaceranger_options"
+ },
{
"$ref": "#/$defs/institutional_config_options"
},
diff --git a/ro-crate-metadata.json b/ro-crate-metadata.json
index 91c8ce1..2591a6f 100644
--- a/ro-crate-metadata.json
+++ b/ro-crate-metadata.json
@@ -23,7 +23,7 @@
"@type": "Dataset",
"creativeWorkStatus": "Stable",
"datePublished": "2025-10-16T13:38:45+00:00",
- "description": "\n \n \n  \n \n
\n \n
\n\n[](https://github.com/codespaces/new/nf-core/sopa)\n[](https://github.com/nf-core/sopa/actions/workflows/nf-test.yml)\n[](https://github.com/nf-core/sopa/actions/workflows/linting.yml)[](https://nf-co.re/sopa/results)[](https://doi.org/10.5281/zenodo.XXXXXXX)\n[](https://www.nf-test.com)\n\n[](https://www.nextflow.io/)\n[](https://github.com/nf-core/tools/releases/tag/3.4.1)\n[](https://docs.conda.io/en/latest/)\n[](https://www.docker.com/)\n[](https://sylabs.io/docs/)\n[](https://cloud.seqera.io/launch?pipeline=https://github.com/nf-core/sopa)\n\n[](https://nfcore.slack.com/channels/sopa)[](https://bsky.app/profile/nf-co.re)[](https://mstdn.science/@nf_core)[](https://www.youtube.com/c/nf-core)\n\n## Introduction\n\n**nf-core/sopa** is a bioinformatics pipeline that ...\n\n\n\n\n\n\n## Usage\n\n> [!NOTE]\n> If you are new to Nextflow and nf-core, please refer to [this page](https://nf-co.re/docs/usage/installation) on how to set-up Nextflow. Make sure to [test your setup](https://nf-co.re/docs/usage/introduction#how-to-run-a-pipeline) with `-profile test` before running the workflow on actual data.\n\n\n\nNow, you can run the pipeline using:\n\n\n\n```bash\nnextflow run nf-core/sopa \\\n -profile \\\n --input samplesheet.csv \\\n --outdir \n```\n\n> [!WARNING]\n> Please provide pipeline parameters via the CLI or Nextflow `-params-file` option. Custom config files including those provided by the `-c` Nextflow option can be used to provide any configuration _**except for parameters**_; see [docs](https://nf-co.re/docs/usage/getting_started/configuration#custom-configuration-files).\n\nFor more details and further functionality, please refer to the [usage documentation](https://nf-co.re/sopa/usage) and the [parameter documentation](https://nf-co.re/sopa/parameters).\n\n## Pipeline output\n\nTo see the results of an example test run with a full size dataset refer to the [results](https://nf-co.re/sopa/results) tab on the nf-core website pipeline page.\nFor more details about the output files and reports, please refer to the\n[output documentation](https://nf-co.re/sopa/output).\n\n## Credits\n\nnf-core/sopa was originally written by Quentin Blampey.\n\nWe thank the following people for their extensive assistance in the development of this pipeline:\n\n\n\n## Contributions and Support\n\nIf you would like to contribute to this pipeline, please see the [contributing guidelines](.github/CONTRIBUTING.md).\n\nFor further information or help, don't hesitate to get in touch on the [Slack `#sopa` channel](https://nfcore.slack.com/channels/sopa) (you can join with [this invite](https://nf-co.re/join/slack)).\n\n## Citations\n\n\n\n\n\n\nAn extensive list of references for the tools used by the pipeline can be found in the [`CITATIONS.md`](CITATIONS.md) file.\n\nYou can cite the `nf-core` publication as follows:\n\n> **The nf-core framework for community-curated bioinformatics pipelines.**\n>\n> Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.\n>\n> _Nat Biotechnol._ 2020 Feb 13. doi: [10.1038/s41587-020-0439-x](https://dx.doi.org/10.1038/s41587-020-0439-x).\n",
+ "description": "\n \n \n \n \n
\n\n[](https://github.com/codespaces/new/nf-core/sopa)\n[](https://github.com/nf-core/sopa/actions/workflows/nf-test.yml)\n[](https://github.com/nf-core/sopa/actions/workflows/linting.yml)[](https://nf-co.re/sopa/results)[](https://doi.org/10.5281/zenodo.XXXXXXX)\n[](https://www.nf-test.com)\n\n[](https://www.nextflow.io/)\n[](https://github.com/nf-core/tools/releases/tag/3.4.1)\n[](https://docs.conda.io/en/latest/)\n[](https://www.docker.com/)\n[](https://sylabs.io/docs/)\n[](https://cloud.seqera.io/launch?pipeline=https://github.com/nf-core/sopa)\n\n[](https://nfcore.slack.com/channels/sopa)[](https://bsky.app/profile/nf-co.re)[](https://mstdn.science/@nf_core)[](https://www.youtube.com/c/nf-core)\n\n## Introduction\n\n**nf-core/sopa** is the Nextflow version of [Sopa](https://github.com/gustaveroussy/sopa). Built on top of [SpatialData](https://github.com/scverse/spatialdata), Sopa enables processing and analyses of spatial omics data with single-cell resolution (spatial transcriptomics or multiplex imaging data) using a standard data structure and output. We currently support the following technologies: Xenium, Visium HD, MERSCOPE, CosMX, PhenoCycler, MACSima, Molecural Cartography, and others. It outputs a `.zarr` directory containing a processed [SpatialData](https://github.com/scverse/spatialdata) object, and a `.explorer` directory for visualization.\n\n> [!WARNING]\n> If you are interested in the main Sopa python package, refer to [this Sopa repository](https://github.com/gustaveroussy/sopa). Else, if you want to use Nextflow, you are in the good place.\n\n\n  \n
\n
\n\n1. (Visium HD only) Raw data processing with Space Ranger\n2. (Optional) Tissue segmentation\n3. Cell segmentation with Cellpose, Baysor, Proseg, Comseg, Stardist, ...\n4. Aggregation, i.e. counting the transcripts inside the cells and/or averaging the channel intensities inside cells\n5. (Optional) Cell-type annotation\n6. User-friendly output creation for visualization and quick analysis\n7. Full [SpatialData](https://github.com/scverse/spatialdata) object export as a `.zarr` directory\n\nAfter running `nf-core/sopa`, you can continue analyzing your `SpatialData` object with [`sopa` as a Python package](https://github.com/gustaveroussy/sopa).\n\n## Usage\n\n> [!NOTE]\n> If you are new to Nextflow and nf-core, please refer to [this page](https://nf-co.re/docs/usage/installation) on how to set-up Nextflow. Make sure to [test your setup](https://nf-co.re/docs/usage/introduction#how-to-run-a-pipeline) with `-profile test` before running the workflow on actual data.\n\nFirst, prepare a samplesheet that lists the `data_path` to each sample data directory (typically, the per-sample output of the Xenium/MERSCOPE/etc, see more info [here](https://gustaveroussy.github.io/sopa/faq/#what-are-the-inputs-of-sopa)). You can optionally add `sample` to provide a name to your output directory, else it will be named based on `data_path`. Here is a samplesheet example:\n\n`samplesheet.csv`:\n\n```csv\nsample,data_path\nSAMPLE1,/path/to/one/merscope_directory\nSAMPLE2,/path/to/one/merscope_directory\n```\n\n> [!WARNING]\n> If you have Visium HD data, the samplesheet will have a different format than the one above. Directly refer to the [usage documentation](https://nf-co.re/sopa/usage) and the [parameter documentation](https://nf-co.re/sopa/parameters).\n\nThen, choose the Sopa parameters. You can find existing Sopa params files [here](https://github.com/gustaveroussy/sopa/tree/main/workflow/config), and follow the [corresponding README instructions](https://github.com/gustaveroussy/sopa/blob/main/workflow/config/README.md) of to get your `-params-file` argument.\n\nNow, you can run the pipeline using:\n\n```bash\nnextflow run nf-core/sopa \\\n -profile \\\n --input samplesheet.csv \\\n -params-file \\\n --outdir \n```\n\n> [!WARNING]\n> Please provide pipeline parameters via the CLI or Nextflow `-params-file` option. Custom config files including those provided by the `-c` Nextflow option can be used to provide any configuration _**except for parameters**_; see [docs](https://nf-co.re/docs/usage/getting_started/configuration#custom-configuration-files).\n\nFor more details and further functionality, please refer to the [usage documentation](https://nf-co.re/sopa/usage) and the [parameter documentation](https://nf-co.re/sopa/parameters).\n\n## Pipeline output\n\nTo see the results of an example test run with a full size dataset refer to the [results](https://nf-co.re/sopa/results) tab on the nf-core website pipeline page.\nFor more details about the output files and reports, please refer to the\n[output documentation](https://nf-co.re/sopa/output).\n\n## Credits\n\nnf-core/sopa was originally written by [Quentin Blampey](https://github.com/quentinblampey) during his work at the following institutions: CentraleSup\u00e9lec, Gustave Roussy Institute, Universit\u00e9 Paris-Saclay, and Cure51.\n\nWe thank the following people for their extensive assistance in the development of this pipeline:\n\n- [Matthias H\u00f6rtenhuber](https://github.com/mashehu)\n- [Kevin Weiss](https://github.com/kweisscure51)\n\n## Contributions and Support\n\nIf you would like to contribute to this pipeline, please see the [contributing guidelines](.github/CONTRIBUTING.md).\n\nFor further information or help, don't hesitate to get in touch on the [Slack `#sopa` channel](https://nfcore.slack.com/channels/sopa) (you can join with [this invite](https://nf-co.re/join/slack)).\n\n## Citations\n\n\n\n\nAn extensive list of references for the tools used by the pipeline can be found in the [`CITATIONS.md`](CITATIONS.md) file.\n\nYou can cite the `sopa` publication as follows:\n\n> Sopa: a technology-invariant pipeline for analyses of image-based spatial omics.\n>\n> Quentin Blampey, Kevin Mulder, Margaux Gardet, Stergios Christodoulidis, Charles-Antoine Dutertre, Fabrice Andr\u00e9, Florent Ginhoux & Paul-Henry Courn\u00e8de.\n>\n> _Nat Commun._ 2024 June 11. doi: [10.1038/s41467-024-48981-z](https://doi.org/10.1038/s41467-024-48981-z)\n\nYou can cite the `nf-core` publication as follows:\n\n> **The nf-core framework for community-curated bioinformatics pipelines.**\n>\n> Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.\n>\n> _Nat Biotechnol._ 2020 Feb 13. doi: [10.1038/s41587-020-0439-x](https://dx.doi.org/10.1038/s41587-020-0439-x).\n",
"hasPart": [
{
"@id": "main.nf"
@@ -40,6 +40,15 @@
{
"@id": "docs/images/"
},
+ {
+ "@id": "modules/"
+ },
+ {
+ "@id": "modules/local/"
+ },
+ {
+ "@id": "modules/nf-core/"
+ },
{
"@id": "workflows/"
},
@@ -115,7 +124,11 @@
},
{
"@id": "main.nf",
- "@type": ["File", "SoftwareSourceCode", "ComputationalWorkflow"],
+ "@type": [
+ "File",
+ "SoftwareSourceCode",
+ "ComputationalWorkflow"
+ ],
"creator": [
{
"@id": "https://orcid.org/0000-0002-3836-2889"
@@ -133,21 +146,30 @@
"spatial-transcriptomics",
"spatialdata"
],
- "license": ["MIT"],
+ "license": [
+ "MIT"
+ ],
"maintainer": [
{
"@id": "https://orcid.org/0000-0002-3836-2889"
}
],
- "name": ["nf-core/sopa"],
+ "name": [
+ "nf-core/sopa"
+ ],
"programmingLanguage": {
"@id": "https://w3id.org/workflowhub/workflow-ro-crate#nextflow"
},
"sdPublisher": {
"@id": "https://nf-co.re/"
},
- "url": ["https://github.com/nf-core/sopa", "https://nf-co.re/sopa/1.0.0/"],
- "version": ["1.0.0"]
+ "url": [
+ "https://github.com/nf-core/sopa",
+ "https://nf-co.re/sopa/1.0.0/"
+ ],
+ "version": [
+ "1.0.0"
+ ]
},
{
"@id": "https://w3id.org/workflowhub/workflow-ro-crate#nextflow",
@@ -212,6 +234,21 @@
"@type": "Dataset",
"description": "Images for the documentation files"
},
+ {
+ "@id": "modules/",
+ "@type": "Dataset",
+ "description": "Modules used by the pipeline"
+ },
+ {
+ "@id": "modules/local/",
+ "@type": "Dataset",
+ "description": "Pipeline-specific modules"
+ },
+ {
+ "@id": "modules/nf-core/",
+ "@type": "Dataset",
+ "description": "nf-core modules"
+ },
{
"@id": "workflows/",
"@type": "Dataset",
@@ -296,8 +333,8 @@
{
"@id": "https://orcid.org/0000-0002-3836-2889",
"@type": "Person",
- "email": "quentin.blampey@gmail.com",
+ "email": "33903498+quentinblampey@users.noreply.github.com",
"name": "Quentin Blampey"
}
]
-}
+}
\ No newline at end of file
diff --git a/subworkflows/local/spaceranger.nf b/subworkflows/local/spaceranger.nf
new file mode 100644
index 0000000..d5c06f7
--- /dev/null
+++ b/subworkflows/local/spaceranger.nf
@@ -0,0 +1,132 @@
+//
+// Raw data processing with Space Ranger
+//
+
+include { UNTAR as SPACERANGER_UNTAR_REFERENCE } from "../../modules/nf-core/untar"
+include { UNTAR as UNTAR_SPACERANGER_INPUT } from "../../modules/nf-core/untar"
+include { SPACERANGER_COUNT } from '../../modules/nf-core/spaceranger/count'
+
+workflow SPACERANGER {
+ take:
+ ch_samplesheet
+
+ main:
+
+ ch_versions = Channel.empty()
+
+ // Space Ranger analysis: --------------------------------------------------
+
+ // Split channel into tarballed and directory inputs
+ ch_spaceranger = ch_samplesheet
+ .map { it -> [it, it.fastq_dir] }
+ .branch {
+ tar: it[1].name.contains(".tar.gz")
+ dir: !it[1].name.contains(".tar.gz")
+ }
+
+ // Extract tarballed inputs
+ UNTAR_SPACERANGER_INPUT(ch_spaceranger.tar)
+ ch_versions = ch_versions.mix(UNTAR_SPACERANGER_INPUT.out.versions)
+
+ // Combine extracted and directory inputs into one channel
+ ch_spaceranger_combined = UNTAR_SPACERANGER_INPUT.out.untar
+ .mix(ch_spaceranger.dir)
+ .map { meta, dir -> meta + [fastq_dir: dir] }
+
+ // Create final meta map and check input existance
+ ch_spaceranger_input = ch_spaceranger_combined.map { create_channel_spaceranger(it) }
+
+
+ //
+ // Reference files
+ //
+ ch_reference = Channel.empty()
+ if (params.spaceranger_reference ==~ /.*\.tar\.gz$/) {
+ ref_file = file(params.spaceranger_reference)
+ SPACERANGER_UNTAR_REFERENCE(
+ [
+ [id: "reference"],
+ ref_file,
+ ]
+ )
+ ch_reference = SPACERANGER_UNTAR_REFERENCE.out.untar.map { _meta, ref -> ref }
+ ch_versions = ch_versions.mix(SPACERANGER_UNTAR_REFERENCE.out.versions)
+ }
+ else {
+ ch_reference = file(params.spaceranger_reference, type: "dir", checkIfExists: true)
+ }
+
+ //
+ // Optional: probe set
+ //
+ ch_probeset = Channel.empty()
+ if (params.spaceranger_probeset) {
+ ch_probeset = file(params.spaceranger_probeset, checkIfExists: true)
+ }
+ else {
+ ch_probeset = []
+ }
+
+ //
+ // Run Space Ranger count
+ //
+ SPACERANGER_COUNT(
+ ch_spaceranger_input,
+ ch_reference,
+ ch_probeset,
+ )
+
+ ch_versions = ch_versions.mix(SPACERANGER_COUNT.out.versions.first())
+
+ emit:
+ sr_dir = SPACERANGER_COUNT.out.outs
+ versions = ch_versions // channel: [ versions.yml ]
+}
+
+
+// Function to get list of [ meta, [ fastq_dir, tissue_hires_image, slide, area ]]
+def create_channel_spaceranger(LinkedHashMap meta) {
+ // Convert a path in `meta` to a file object and return it. If `key` is not contained in `meta`
+ // return an empty list which is recognized as 'no file' by nextflow.
+ def get_file_from_meta = { key ->
+ def v = meta[key]
+ return v ? file(v) : []
+ }
+
+ def slide = meta.remove("slide")
+ def area = meta.remove("area")
+ def fastq_dir = meta.remove("fastq_dir")
+ def fastq_files = file("${fastq_dir}/${meta['id']}*.fastq.gz")
+ def manual_alignment = get_file_from_meta("manual_alignment")
+ def slidefile = get_file_from_meta("slidefile")
+ def image = get_file_from_meta("image")
+ def cytaimage = get_file_from_meta("cytaimage")
+ def colorizedimage = get_file_from_meta("colorizedimage")
+ def darkimage = get_file_from_meta("darkimage")
+
+ if (!fastq_files.size()) {
+ error("No `fastq_dir` specified or no samples found in folder.")
+ }
+
+ // Check for existance of optional files
+ def optional_files = [
+ 'manual_alignment': manual_alignment,
+ 'slidefile': slidefile,
+ 'image': image,
+ 'cytaimage': cytaimage,
+ 'colorizedimage': colorizedimage,
+ 'darkimage': darkimage,
+ ]
+ optional_files.each { k, f ->
+ if (f && !f.exists()) {
+ error("File for `${k}` is specified, but does not exist: ${f}.")
+ }
+ }
+
+ // Check that at least one type of image is specified
+ if (!(image || cytaimage || colorizedimage || darkimage)) {
+ error("Need to specify at least one of 'image', 'cytaimage', 'colorizedimage', or 'darkimage' in the samplesheet")
+ }
+
+ return [meta, fastq_files, image, slide, area, cytaimage, darkimage, colorizedimage, manual_alignment, slidefile]
+}
diff --git a/subworkflows/local/utils_nfcore_sopa_pipeline/main.nf b/subworkflows/local/utils_nfcore_sopa_pipeline/main.nf
index 19f13ac..93dcdaf 100644
--- a/subworkflows/local/utils_nfcore_sopa_pipeline/main.nf
+++ b/subworkflows/local/utils_nfcore_sopa_pipeline/main.nf
@@ -25,11 +25,10 @@ include { UTILS_NEXTFLOW_PIPELINE } from '../../nf-core/utils_nextflow_pipelin
*/
workflow PIPELINE_INITIALISATION {
-
take:
- version // boolean: Display version and exit
- validate_params // boolean: Boolean whether to validate parameters against the schema at runtime
- monochrome_logs // boolean: Do not use coloured log outputs
+ version // boolean: Display version and exit
+ validate_params // boolean: Boolean whether to validate parameters against the schema at runtime
+ monochrome_logs // boolean: Do not use coloured log outputs
nextflow_cli_args // array: List of positional nextflow CLI args
outdir // string: The output directory where the results will be saved
input // string: Path to input samplesheet
@@ -44,11 +43,11 @@ workflow PIPELINE_INITIALISATION {
//
// Print version and exit if required and dump pipeline parameters to JSON file
//
- UTILS_NEXTFLOW_PIPELINE (
+ UTILS_NEXTFLOW_PIPELINE(
version,
true,
outdir,
- workflow.profile.tokenize(',').intersect(['conda', 'mamba']).size() >= 1
+ workflow.profile.tokenize(',').intersect(['conda', 'mamba']).size() >= 1,
)
//
@@ -88,7 +87,7 @@ workflow PIPELINE_INITIALISATION {
//
// Check config provided to the pipeline
//
- UTILS_NFCORE_PIPELINE (
+ UTILS_NFCORE_PIPELINE(
nextflow_cli_args
)
@@ -96,29 +95,53 @@ workflow PIPELINE_INITIALISATION {
// Create channel from input file provided through params.input
//
- Channel
- .fromList(samplesheetToList(params.input, "${projectDir}/assets/schema_input.json"))
- .map {
- meta, fastq_1, fastq_2 ->
- if (!fastq_2) {
- return [ meta.id, meta + [ single_end:true ], [ fastq_1 ] ]
- } else {
- return [ meta.id, meta + [ single_end:false ], [ fastq_1, fastq_2 ] ]
+ Channel.fromList(samplesheetToList(params.input, "${projectDir}/assets/schema_input.json"))
+ .map { meta, data_path ->
+ if (!meta.fastq_dir) {
+ if (!data_path) {
+ error("The `data_path` must be provided (path to the raw inputs), except when running on Visium HD data (in that case, the `fastq_dir` is required)")
+ }
+
+ if (!meta.sample) {
+ meta.sample = file(data_path).baseName
+ }
+
+ meta.data_dir = data_path
+ }
+ else {
+ // spaceranger output directory
+ meta.data_dir = "outs"
+
+ if (!meta.sample) {
+ error("The `sample` column must be provided when running on Visium HD data")
+ }
+
+ if (!meta.id) {
+ meta.id = meta.sample
+ }
+
+ if (!meta.image) {
+ error("The `image` column (full resolution image) must be provided when running Sopa on Visium HD data - it is required for the cell segmentation")
}
+ }
+ meta.sdata_dir = "${meta.sample}.zarr"
+ meta.explorer_dir = "${meta.sample}.explorer"
+
+ return meta
}
- .groupTuple()
.map { samplesheet ->
validateInputSamplesheet(samplesheet)
}
- .map {
- meta, fastqs ->
- return [ meta, fastqs.flatten() ]
- }
.set { ch_samplesheet }
+ //
+ // Sopa params validation
+ //
+ validateParams(params)
+
emit:
samplesheet = ch_samplesheet
- versions = ch_versions
+ versions = ch_versions
}
/*
@@ -128,14 +151,13 @@ workflow PIPELINE_INITIALISATION {
*/
workflow PIPELINE_COMPLETION {
-
take:
- email // string: email address
- email_on_fail // string: email address sent on pipeline failure
+ email // string: email address
+ email_on_fail // string: email address sent on pipeline failure
plaintext_email // boolean: Send plain-text email instead of HTML
- outdir // path: Path to output directory where results will be published
+ outdir // path: Path to output directory where results will be published
monochrome_logs // boolean: Disable ANSI colour codes in log output
- hook_url // string: hook URL for notifications
+ hook_url // string: hook URL for notifications
main:
summary_params = paramsSummaryMap(workflow, parameters_schema: "nextflow_schema.json")
@@ -152,7 +174,7 @@ workflow PIPELINE_COMPLETION {
plaintext_email,
outdir,
monochrome_logs,
- []
+ [],
)
}
@@ -163,7 +185,7 @@ workflow PIPELINE_COMPLETION {
}
workflow.onError {
- log.error "Pipeline failed. Please refer to troubleshooting docs: https://nf-co.re/docs/usage/troubleshooting"
+ log.error("Pipeline failed. Please refer to troubleshooting docs: https://nf-co.re/docs/usage/troubleshooting")
}
}
@@ -177,37 +199,36 @@ workflow PIPELINE_COMPLETION {
// Validate channels from input samplesheet
//
def validateInputSamplesheet(input) {
- def (metas, fastqs) = input[1..2]
-
- // Check that multiple runs of the same sample are of the same datatype i.e. single-end / paired-end
- def endedness_ok = metas.collect{ meta -> meta.single_end }.unique().size == 1
- if (!endedness_ok) {
- error("Please check input samplesheet -> Multiple runs of a sample must be of the same datatype i.e. single-end or paired-end: ${metas[0].id}")
- }
-
- return [ metas[0], fastqs ]
+ return input
}
//
// Generate methods description for MultiQC
//
def toolCitationText() {
- // TODO nf-core: Optionally add in-text citation tools to this list.
- // Can use ternary operators to dynamically construct based conditions, e.g. params["run_xyz"] ? "Tool (Foo et al. 2023)" : "",
- // Uncomment function in methodsDescriptionText to render in MultiQC report
def citation_text = [
- "Tools used in the workflow included:",
- "."
- ].join(' ').trim()
+ "Tools used in the workflow included:",
+ "Sopa (Blampey et al. 2024),",
+ "AnnData (Virshup et al. 2021),",
+ "Scanpy (Wolf et al. 2018),",
+ "Space Ranger (10x Genomics)",
+ "SpatialData (Marconato et al. 2023) and",
+ ].join(' ').trim()
return citation_text
}
def toolBibliographyText() {
- // TODO nf-core: Optionally add bibliographic entries to this list.
- // Can use ternary operators to dynamically construct based conditions, e.g. params["run_xyz"] ? "Author (2023) Pub name, Journal, DOI" : "",
- // Uncomment function in methodsDescriptionText to render in MultiQC report
def reference_text = [
- ].join(' ').trim()
+ 'Di Tommaso, P., Chatzou, M., Floden, E. W., Barja, P. P., Palumbo, E., & Notredame, C. (2017). Nextflow enables reproducible computational workflows. Nature Biotechnology, 35(4), 316-319. doi: 10.1038/nbt.3820',
+ 'Ewels, P. A., Peltzer, A., Fillinger, S., Patel, H., Alneberg, J., Wilm, A., Garcia, M. U., Di Tommaso, P., & Nahnsen, S. (2020). The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology, 38(3), 276-278. doi: 10.1038/s41587-020-0439-x',
+ 'Grüning, B., Dale, R., Sjödin, A., Chapman, B. A., Rowe, J., Tomkins-Tinch, C. H., Valieris, R., Köster, J., & Bioconda Team. (2018). Bioconda: sustainable and comprehensive software distribution for the life sciences. Nature Methods, 15(7), 475–476. doi: 10.1038/s41592-018-0046-7',
+ 'da Veiga Leprevost, F., Grüning, B. A., Alves Aflitos, S., Röst, H. L., Uszkoreit, J., Barsnes, H., Vaudel, M., Moreno, P., Gatto, L., Weber, J., Bai, M., Jimenez, R. C., Sachsenberg, T., Pfeuffer, J., Vera Alvarez, R., Griss, J., Nesvizhskii, A. I., & Perez-Riverol, Y. (2017). BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics (Oxford, England), 33(16), 2580–2582. doi: 10.1093/bioinformatics/btx192',
+ 'Quentin Blampey, Kevin Mulder, Margaux Gardet, Stergios Christodoulidis, Charles-Antoine Dutertre, Fabrice André, Florent Ginhoux & Paul-Henry Cournède. Sopa: a technology-invariant pipeline for analyses of image-based spatial omics. Nat Commun 2024 June 11. doi: 10.1038/s41587-020-0439-x',
+ 'Virshup I, Rybakov S, Theis FJ, Angerer P, Wolf FA. bioRxiv 2021.12.16.473007. doi: 10.1101/2021.12.16.473007',
+ 'Wolf F, Angerer P, Theis F. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15 (2018). doi: 10.1186/s13059-017-1382-0',
+ '10x Genomics Space Ranger 2.1.0 [Online]: 10xgenomics.com/support/software/space-ranger',
+ 'Marconato L, Palla G, Yamauchi K, Virshup I, Heidari E, Treis T, Toth M, Shrestha R, Vöhringer H, Huber W, Gerstung M, Moore J, Theis F, Stegle O. SpatialData: an open and universal data framework for spatial omics. bioRxiv 2023.05.05.539647; doi: 10.1101/2023.05.05.539647',
+ ].join(' ').trim()
return reference_text
}
@@ -229,22 +250,66 @@ def methodsDescriptionText(mqc_methods_yaml) {
temp_doi_ref += "(doi: ${doi_ref.replace("https://doi.org/", "").replace(" ", "")}), "
}
meta["doi_text"] = temp_doi_ref.substring(0, temp_doi_ref.length() - 2)
- } else meta["doi_text"] = ""
+ }
+ else {
+ meta["doi_text"] = ""
+ }
meta["nodoi_text"] = meta.manifest_map.doi ? "" : "If available, make sure to update the text to include the Zenodo DOI of version of the pipeline used. "
// Tool references
- meta["tool_citations"] = ""
- meta["tool_bibliography"] = ""
-
- // TODO nf-core: Only uncomment below if logic in toolCitationText/toolBibliographyText has been filled!
- // meta["tool_citations"] = toolCitationText().replaceAll(", \\.", ".").replaceAll("\\. \\.", ".").replaceAll(", \\.", ".")
- // meta["tool_bibliography"] = toolBibliographyText()
+ meta["tool_citations"] = toolCitationText().replaceAll(", \\.", ".").replaceAll("\\. \\.", ".").replaceAll(", \\.", ".")
+ meta["tool_bibliography"] = toolBibliographyText()
def methods_text = mqc_methods_yaml.text
- def engine = new groovy.text.SimpleTemplateEngine()
+ def engine = new groovy.text.SimpleTemplateEngine()
def description_html = engine.createTemplate(methods_text).make(meta)
return description_html.toString()
}
+
+def validateParams(params) {
+ def TRANSCRIPT_BASED_METHODS = ['proseg', 'baysor', 'comseg']
+ def STAINING_BASED_METHODS = ['stardist', 'cellpose']
+
+ // top-level checks
+ assert params.read instanceof Map && params.read.containsKey('technology') : "Provide a 'read.technology' key"
+ assert params.containsKey('segmentation') : "Provide a 'segmentation' section"
+
+ // backward compatibility
+ TRANSCRIPT_BASED_METHODS.each { m ->
+ if (params.segmentation?.get(m)?.containsKey('cell_key')) {
+ println("Deprecated 'cell_key' → using 'prior_shapes_key' instead.")
+ params.segmentation[m].prior_shapes_key = params.segmentation[m].cell_key
+ params.segmentation[m].remove('cell_key')
+ }
+ }
+ if (params.aggregate?.containsKey('average_intensities')) {
+ println("Deprecated 'average_intensities' → using 'aggregate_channels' instead.")
+ params.aggregate.aggregate_channels = params.aggregate.average_intensities
+ params.aggregate.remove('average_intensities')
+ }
+
+ // check segmentation methods
+ assert params.segmentation : "Provide at least one segmentation method"
+ assert TRANSCRIPT_BASED_METHODS.count { params.segmentation.containsKey(it) } <= 1 : "Only one of ${TRANSCRIPT_BASED_METHODS} may be used"
+ assert STAINING_BASED_METHODS.count { params.segmentation.containsKey(it) } <= 1 : "Only one of ${STAINING_BASED_METHODS} may be used"
+ if (params.segmentation.containsKey('stardist')) {
+ assert TRANSCRIPT_BASED_METHODS.every { !params.segmentation.containsKey(it) } : "'stardist' cannot be combined with transcript-based methods"
+ }
+
+ // check prior shapes key
+ TRANSCRIPT_BASED_METHODS.each { m ->
+ if (params.segmentation.containsKey(m) && params.segmentation.containsKey('cellpose')) {
+ params.segmentation[m].prior_shapes_key = 'cellpose_boundaries'
+ }
+ }
+
+ // check annotation method
+ if (params.annotation && params.annotation.method == "tangram") {
+ assert params.annotation.args.containsKey('sc_reference_path') : "Provide 'annotation.args.sc_reference_path' for the tangram annotation method"
+ }
+
+ return params
+}
diff --git a/tests/.nftignore b/tests/.nftignore
index 73eb92f..7d01b42 100644
--- a/tests/.nftignore
+++ b/tests/.nftignore
@@ -1,2 +1,19 @@
.DS_Store
pipeline_info/*.{html,json,txt,yml}
+**/part.**
+**.zarr.zip
+**/adata.h5ad
+**/analysis_summary.html
+**/experiment.xenium
+**/morphology.ome.tif
+**/shapes.parquet
+**/.zattrs
+**/.zarray
+**/0
+**/1
+**/2
+**/3
+**/zmetadata
+**/.sopa_cache
+**/.sopa_cache/**
+**/table/**
diff --git a/tests/baysor.nf.test b/tests/baysor.nf.test
new file mode 100644
index 0000000..946801b
--- /dev/null
+++ b/tests/baysor.nf.test
@@ -0,0 +1,36 @@
+nextflow_pipeline {
+
+ name "Test pipeline"
+ script "../main.nf"
+ profile "test_baysor"
+ tag "pipeline"
+
+ test("-profile test_baysor") {
+
+ when {
+ params {
+ outdir = "$outputDir"
+ }
+ }
+
+ then {
+ // stable_name: All files + folders in ${params.outdir}/ with a stable name
+ def stable_name = getAllFilesFromDir(params.outdir, relative: true, includeDir: true, ignore: ['pipeline_info/*.{html,json,txt}'])
+ // stable_path: All files in ${params.outdir}/ with stable content
+ def stable_path = getAllFilesFromDir(params.outdir, ignoreFile: 'tests/.nftignore')
+ assertAll(
+ { assert workflow.success},
+ { assert snapshot(
+ // Number of successful tasks
+ workflow.trace.succeeded().size(),
+ // pipeline versions.yml file for multiqc from which Nextflow version is removed because we test pipelines on multiple Nextflow versions
+ removeNextflowVersion("$outputDir/pipeline_info/nf_core_sopa_software_mqc_versions.yml"),
+ // All stable path name, with a relative path
+ stable_name,
+ // All files with stable contents
+ stable_path
+ ).match() }
+ )
+ }
+ }
+}
diff --git a/tests/baysor.nf.test.snap b/tests/baysor.nf.test.snap
new file mode 100644
index 0000000..7c58ec0
--- /dev/null
+++ b/tests/baysor.nf.test.snap
@@ -0,0 +1,360 @@
+{
+ "-profile test_baysor": {
+ "content": [
+ 9,
+ {
+ "RESOLVE_BAYSOR": {
+ "sopa": "2.1.8",
+ "baysor": "0.7.1"
+ },
+ "TANGRAM_ANNOTATION": {
+ "sopa": "2.1.8",
+ "tangram": "1.0.4"
+ },
+ "TO_SPATIALDATA": {
+ "sopa": "2.1.8",
+ "spatialdata": "0.5.0",
+ "spatialdata_io": "0.3.0"
+ },
+ "Workflow": {
+ "nf-core/sopa": "v1.0.0"
+ }
+ },
+ [
+ "pipeline_info",
+ "pipeline_info/nf_core_sopa_software_mqc_versions.yml",
+ "sample_name.explorer",
+ "sample_name.explorer/adata.h5ad",
+ "sample_name.explorer/analysis.zarr.zip",
+ "sample_name.explorer/analysis_summary.html",
+ "sample_name.explorer/cell_feature_matrix.zarr.zip",
+ "sample_name.explorer/cells.zarr.zip",
+ "sample_name.explorer/experiment.xenium",
+ "sample_name.explorer/morphology.ome.tif",
+ "sample_name.explorer/transcripts.zarr.zip",
+ "sample_name.zarr",
+ "sample_name.zarr/.zattrs",
+ "sample_name.zarr/.zgroup",
+ "sample_name.zarr/images",
+ "sample_name.zarr/images/.zgroup",
+ "sample_name.zarr/images/he_image",
+ "sample_name.zarr/images/he_image/.zattrs",
+ "sample_name.zarr/images/he_image/.zgroup",
+ "sample_name.zarr/images/he_image/0",
+ "sample_name.zarr/images/he_image/0/.zarray",
+ "sample_name.zarr/images/he_image/0/0",
+ "sample_name.zarr/images/he_image/0/0/0",
+ "sample_name.zarr/images/he_image/0/0/0/0",
+ "sample_name.zarr/images/he_image/0/0/0/1",
+ "sample_name.zarr/images/he_image/0/0/0/2",
+ "sample_name.zarr/images/he_image/0/0/0/3",
+ "sample_name.zarr/images/he_image/0/0/1",
+ "sample_name.zarr/images/he_image/0/0/1/0",
+ "sample_name.zarr/images/he_image/0/0/1/1",
+ "sample_name.zarr/images/he_image/0/0/1/2",
+ "sample_name.zarr/images/he_image/0/0/1/3",
+ "sample_name.zarr/images/he_image/0/0/2",
+ "sample_name.zarr/images/he_image/0/0/2/0",
+ "sample_name.zarr/images/he_image/0/0/2/1",
+ "sample_name.zarr/images/he_image/0/0/2/2",
+ "sample_name.zarr/images/he_image/0/0/2/3",
+ "sample_name.zarr/images/he_image/0/0/3",
+ "sample_name.zarr/images/he_image/0/0/3/0",
+ "sample_name.zarr/images/he_image/0/0/3/1",
+ "sample_name.zarr/images/he_image/0/0/3/2",
+ "sample_name.zarr/images/he_image/0/0/3/3",
+ "sample_name.zarr/images/he_image/1",
+ "sample_name.zarr/images/he_image/1/.zarray",
+ "sample_name.zarr/images/he_image/1/0",
+ "sample_name.zarr/images/he_image/1/0/0",
+ "sample_name.zarr/images/he_image/1/0/0/0",
+ "sample_name.zarr/images/he_image/1/0/0/1",
+ "sample_name.zarr/images/he_image/1/0/1",
+ "sample_name.zarr/images/he_image/1/0/1/0",
+ "sample_name.zarr/images/he_image/1/0/1/1",
+ "sample_name.zarr/images/he_image/2",
+ "sample_name.zarr/images/he_image/2/.zarray",
+ "sample_name.zarr/images/he_image/2/0",
+ "sample_name.zarr/images/he_image/2/0/0",
+ "sample_name.zarr/images/he_image/2/0/0/0",
+ "sample_name.zarr/images/image",
+ "sample_name.zarr/images/image/.zattrs",
+ "sample_name.zarr/images/image/.zgroup",
+ "sample_name.zarr/images/image/0",
+ "sample_name.zarr/images/image/0/.zarray",
+ "sample_name.zarr/images/image/0/0",
+ "sample_name.zarr/images/image/0/0/0",
+ "sample_name.zarr/images/image/0/0/0/0",
+ "sample_name.zarr/images/image/0/0/0/1",
+ "sample_name.zarr/images/image/0/0/1",
+ "sample_name.zarr/images/image/0/0/1/0",
+ "sample_name.zarr/images/image/0/0/1/1",
+ "sample_name.zarr/images/image/0/1",
+ "sample_name.zarr/images/image/0/1/0",
+ "sample_name.zarr/images/image/0/1/0/0",
+ "sample_name.zarr/images/image/0/1/0/1",
+ "sample_name.zarr/images/image/0/1/1",
+ "sample_name.zarr/images/image/0/1/1/0",
+ "sample_name.zarr/images/image/0/1/1/1",
+ "sample_name.zarr/images/image/0/2",
+ "sample_name.zarr/images/image/0/2/0",
+ "sample_name.zarr/images/image/0/2/0/0",
+ "sample_name.zarr/images/image/0/2/0/1",
+ "sample_name.zarr/images/image/0/2/1",
+ "sample_name.zarr/images/image/0/2/1/0",
+ "sample_name.zarr/images/image/0/2/1/1",
+ "sample_name.zarr/images/image/0/3",
+ "sample_name.zarr/images/image/0/3/0",
+ "sample_name.zarr/images/image/0/3/0/0",
+ "sample_name.zarr/images/image/0/3/0/1",
+ "sample_name.zarr/images/image/0/3/1",
+ "sample_name.zarr/images/image/0/3/1/0",
+ "sample_name.zarr/images/image/0/3/1/1",
+ "sample_name.zarr/points",
+ "sample_name.zarr/points/.zgroup",
+ "sample_name.zarr/points/transcripts",
+ "sample_name.zarr/points/transcripts/.zattrs",
+ "sample_name.zarr/points/transcripts/.zgroup",
+ "sample_name.zarr/points/transcripts/points.parquet",
+ "sample_name.zarr/points/transcripts/points.parquet/part.0.parquet",
+ "sample_name.zarr/shapes",
+ "sample_name.zarr/shapes/.zgroup",
+ "sample_name.zarr/shapes/baysor_boundaries",
+ "sample_name.zarr/shapes/baysor_boundaries/.zattrs",
+ "sample_name.zarr/shapes/baysor_boundaries/.zgroup",
+ "sample_name.zarr/shapes/baysor_boundaries/shapes.parquet",
+ "sample_name.zarr/shapes/cells",
+ "sample_name.zarr/shapes/cells/.zattrs",
+ "sample_name.zarr/shapes/cells/.zgroup",
+ "sample_name.zarr/shapes/cells/shapes.parquet",
+ "sample_name.zarr/shapes/transcripts_patches",
+ "sample_name.zarr/shapes/transcripts_patches/.zattrs",
+ "sample_name.zarr/shapes/transcripts_patches/.zgroup",
+ "sample_name.zarr/shapes/transcripts_patches/shapes.parquet",
+ "sample_name.zarr/tables",
+ "sample_name.zarr/tables/.zgroup",
+ "sample_name.zarr/tables/table",
+ "sample_name.zarr/tables/table/.zattrs",
+ "sample_name.zarr/tables/table/.zgroup",
+ "sample_name.zarr/tables/table/X",
+ "sample_name.zarr/tables/table/X/.zattrs",
+ "sample_name.zarr/tables/table/X/.zgroup",
+ "sample_name.zarr/tables/table/X/data",
+ "sample_name.zarr/tables/table/X/data/.zarray",
+ "sample_name.zarr/tables/table/X/data/0",
+ "sample_name.zarr/tables/table/X/indices",
+ "sample_name.zarr/tables/table/X/indices/.zarray",
+ "sample_name.zarr/tables/table/X/indices/0",
+ "sample_name.zarr/tables/table/X/indptr",
+ "sample_name.zarr/tables/table/X/indptr/.zarray",
+ "sample_name.zarr/tables/table/X/indptr/0",
+ "sample_name.zarr/tables/table/layers",
+ "sample_name.zarr/tables/table/layers/.zattrs",
+ "sample_name.zarr/tables/table/layers/.zgroup",
+ "sample_name.zarr/tables/table/obs",
+ "sample_name.zarr/tables/table/obs/.zattrs",
+ "sample_name.zarr/tables/table/obs/.zgroup",
+ "sample_name.zarr/tables/table/obs/_index",
+ "sample_name.zarr/tables/table/obs/_index/.zarray",
+ "sample_name.zarr/tables/table/obs/_index/.zattrs",
+ "sample_name.zarr/tables/table/obs/_index/0",
+ "sample_name.zarr/tables/table/obs/area",
+ "sample_name.zarr/tables/table/obs/area/.zarray",
+ "sample_name.zarr/tables/table/obs/area/.zattrs",
+ "sample_name.zarr/tables/table/obs/area/0",
+ "sample_name.zarr/tables/table/obs/avg_assignment_confidence",
+ "sample_name.zarr/tables/table/obs/avg_assignment_confidence/.zarray",
+ "sample_name.zarr/tables/table/obs/avg_assignment_confidence/.zattrs",
+ "sample_name.zarr/tables/table/obs/avg_assignment_confidence/0",
+ "sample_name.zarr/tables/table/obs/avg_confidence",
+ "sample_name.zarr/tables/table/obs/avg_confidence/.zarray",
+ "sample_name.zarr/tables/table/obs/avg_confidence/.zattrs",
+ "sample_name.zarr/tables/table/obs/avg_confidence/0",
+ "sample_name.zarr/tables/table/obs/baysor_area",
+ "sample_name.zarr/tables/table/obs/baysor_area/.zarray",
+ "sample_name.zarr/tables/table/obs/baysor_area/.zattrs",
+ "sample_name.zarr/tables/table/obs/baysor_area/0",
+ "sample_name.zarr/tables/table/obs/cell_id",
+ "sample_name.zarr/tables/table/obs/cell_id/.zarray",
+ "sample_name.zarr/tables/table/obs/cell_id/.zattrs",
+ "sample_name.zarr/tables/table/obs/cell_id/0",
+ "sample_name.zarr/tables/table/obs/cluster",
+ "sample_name.zarr/tables/table/obs/cluster/.zarray",
+ "sample_name.zarr/tables/table/obs/cluster/.zattrs",
+ "sample_name.zarr/tables/table/obs/cluster/0",
+ "sample_name.zarr/tables/table/obs/ct",
+ "sample_name.zarr/tables/table/obs/ct/.zarray",
+ "sample_name.zarr/tables/table/obs/ct/.zattrs",
+ "sample_name.zarr/tables/table/obs/ct/0",
+ "sample_name.zarr/tables/table/obs/density",
+ "sample_name.zarr/tables/table/obs/density/.zarray",
+ "sample_name.zarr/tables/table/obs/density/.zattrs",
+ "sample_name.zarr/tables/table/obs/density/0",
+ "sample_name.zarr/tables/table/obs/elongation",
+ "sample_name.zarr/tables/table/obs/elongation/.zarray",
+ "sample_name.zarr/tables/table/obs/elongation/.zattrs",
+ "sample_name.zarr/tables/table/obs/elongation/0",
+ "sample_name.zarr/tables/table/obs/lifespan",
+ "sample_name.zarr/tables/table/obs/lifespan/.zarray",
+ "sample_name.zarr/tables/table/obs/lifespan/.zattrs",
+ "sample_name.zarr/tables/table/obs/lifespan/0",
+ "sample_name.zarr/tables/table/obs/max_cluster_frac",
+ "sample_name.zarr/tables/table/obs/max_cluster_frac/.zarray",
+ "sample_name.zarr/tables/table/obs/max_cluster_frac/.zattrs",
+ "sample_name.zarr/tables/table/obs/max_cluster_frac/0",
+ "sample_name.zarr/tables/table/obs/n_transcripts",
+ "sample_name.zarr/tables/table/obs/n_transcripts/.zarray",
+ "sample_name.zarr/tables/table/obs/n_transcripts/.zattrs",
+ "sample_name.zarr/tables/table/obs/n_transcripts/0",
+ "sample_name.zarr/tables/table/obs/region",
+ "sample_name.zarr/tables/table/obs/region/.zattrs",
+ "sample_name.zarr/tables/table/obs/region/.zgroup",
+ "sample_name.zarr/tables/table/obs/region/categories",
+ "sample_name.zarr/tables/table/obs/region/categories/.zarray",
+ "sample_name.zarr/tables/table/obs/region/categories/.zattrs",
+ "sample_name.zarr/tables/table/obs/region/categories/0",
+ "sample_name.zarr/tables/table/obs/region/codes",
+ "sample_name.zarr/tables/table/obs/region/codes/.zarray",
+ "sample_name.zarr/tables/table/obs/region/codes/.zattrs",
+ "sample_name.zarr/tables/table/obs/region/codes/0",
+ "sample_name.zarr/tables/table/obs/slide",
+ "sample_name.zarr/tables/table/obs/slide/.zattrs",
+ "sample_name.zarr/tables/table/obs/slide/.zgroup",
+ "sample_name.zarr/tables/table/obs/slide/categories",
+ "sample_name.zarr/tables/table/obs/slide/categories/.zarray",
+ "sample_name.zarr/tables/table/obs/slide/categories/.zattrs",
+ "sample_name.zarr/tables/table/obs/slide/categories/0",
+ "sample_name.zarr/tables/table/obs/slide/codes",
+ "sample_name.zarr/tables/table/obs/slide/codes/.zarray",
+ "sample_name.zarr/tables/table/obs/slide/codes/.zattrs",
+ "sample_name.zarr/tables/table/obs/slide/codes/0",
+ "sample_name.zarr/tables/table/obs/x",
+ "sample_name.zarr/tables/table/obs/x/.zarray",
+ "sample_name.zarr/tables/table/obs/x/.zattrs",
+ "sample_name.zarr/tables/table/obs/x/0",
+ "sample_name.zarr/tables/table/obs/y",
+ "sample_name.zarr/tables/table/obs/y/.zarray",
+ "sample_name.zarr/tables/table/obs/y/.zattrs",
+ "sample_name.zarr/tables/table/obs/y/0",
+ "sample_name.zarr/tables/table/obsm",
+ "sample_name.zarr/tables/table/obsm/.zattrs",
+ "sample_name.zarr/tables/table/obsm/.zgroup",
+ "sample_name.zarr/tables/table/obsm/intensities",
+ "sample_name.zarr/tables/table/obsm/intensities/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/.zgroup",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20/0",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3/0",
+ "sample_name.zarr/tables/table/obsm/intensities/CK",
+ "sample_name.zarr/tables/table/obsm/intensities/CK/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/CK/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/CK/0",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI/0",
+ "sample_name.zarr/tables/table/obsm/intensities/_index",
+ "sample_name.zarr/tables/table/obsm/intensities/_index/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/_index/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/_index/0",
+ "sample_name.zarr/tables/table/obsm/spatial",
+ "sample_name.zarr/tables/table/obsm/spatial/.zarray",
+ "sample_name.zarr/tables/table/obsm/spatial/.zattrs",
+ "sample_name.zarr/tables/table/obsm/spatial/0",
+ "sample_name.zarr/tables/table/obsm/spatial/0/0",
+ "sample_name.zarr/tables/table/obsm/tangram_pred",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/.zattrs",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/.zgroup",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/B cell",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/B cell/.zarray",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/B cell/.zattrs",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/B cell/0",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/T cell",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/T cell/.zarray",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/T cell/.zattrs",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/T cell/0",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/Tumor",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/Tumor/.zarray",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/Tumor/.zattrs",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/Tumor/0",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/_index",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/_index/.zarray",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/_index/.zattrs",
+ "sample_name.zarr/tables/table/obsm/tangram_pred/_index/0",
+ "sample_name.zarr/tables/table/obsp",
+ "sample_name.zarr/tables/table/obsp/.zattrs",
+ "sample_name.zarr/tables/table/obsp/.zgroup",
+ "sample_name.zarr/tables/table/uns",
+ "sample_name.zarr/tables/table/uns/.zattrs",
+ "sample_name.zarr/tables/table/uns/.zgroup",
+ "sample_name.zarr/tables/table/uns/sopa_attrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/.zattrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/.zgroup",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/cell_types",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/cell_types/.zarray",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/cell_types/.zattrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/cell_types/0",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities/.zarray",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities/.zattrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities/0",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts/.zarray",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts/.zattrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts/0",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/.zgroup",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key/.zarray",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key/0",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region/.zarray",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region/0",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key/.zarray",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key/0",
+ "sample_name.zarr/tables/table/var",
+ "sample_name.zarr/tables/table/var/.zattrs",
+ "sample_name.zarr/tables/table/var/.zgroup",
+ "sample_name.zarr/tables/table/var/Name",
+ "sample_name.zarr/tables/table/var/Name/.zarray",
+ "sample_name.zarr/tables/table/var/Name/.zattrs",
+ "sample_name.zarr/tables/table/var/Name/0",
+ "sample_name.zarr/tables/table/varm",
+ "sample_name.zarr/tables/table/varm/.zattrs",
+ "sample_name.zarr/tables/table/varm/.zgroup",
+ "sample_name.zarr/tables/table/varp",
+ "sample_name.zarr/tables/table/varp/.zattrs",
+ "sample_name.zarr/tables/table/varp/.zgroup",
+ "sample_name.zarr/zmetadata"
+ ],
+ [
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab"
+ ]
+ ],
+ "meta": {
+ "nf-test": "0.9.2",
+ "nextflow": "25.04.6"
+ },
+ "timestamp": "2025-10-15T11:14:18.110413"
+ }
+}
\ No newline at end of file
diff --git a/tests/cellpose.nf.test b/tests/cellpose.nf.test
new file mode 100644
index 0000000..6fc0b0e
--- /dev/null
+++ b/tests/cellpose.nf.test
@@ -0,0 +1,36 @@
+nextflow_pipeline {
+
+ name "Test pipeline"
+ script "../main.nf"
+ profile "test_cellpose"
+ tag "pipeline"
+
+ test("-profile test_cellpose") {
+
+ when {
+ params {
+ outdir = "$outputDir"
+ }
+ }
+
+ then {
+ // stable_name: All files + folders in ${params.outdir}/ with a stable name
+ def stable_name = getAllFilesFromDir(params.outdir, relative: true, includeDir: true, ignore: ['pipeline_info/*.{html,json,txt}', '**/.sopa_cache', '**/.sopa_cache/**'])
+ // stable_path: All files in ${params.outdir}/ with stable content
+ def stable_path = getAllFilesFromDir(params.outdir, ignoreFile: 'tests/.nftignore')
+ assertAll(
+ { assert workflow.success},
+ { assert snapshot(
+ // Number of successful tasks
+ workflow.trace.succeeded().size(),
+ // pipeline versions.yml file for multiqc from which Nextflow version is removed because we test pipelines on multiple Nextflow versions
+ removeNextflowVersion("$outputDir/pipeline_info/nf_core_sopa_software_mqc_versions.yml"),
+ // All stable path name, with a relative path
+ stable_name,
+ // All files with stable contents
+ stable_path
+ ).match() }
+ )
+ }
+ }
+}
diff --git a/tests/cellpose.nf.test.snap b/tests/cellpose.nf.test.snap
new file mode 100644
index 0000000..9a92b79
--- /dev/null
+++ b/tests/cellpose.nf.test.snap
@@ -0,0 +1,285 @@
+{
+ "-profile test_cellpose": {
+ "content": [
+ 8,
+ {
+ "RESOLVE_CELLPOSE": {
+ "sopa": "2.1.8",
+ "cellpose": "4.0.7"
+ },
+ "TO_SPATIALDATA": {
+ "sopa": "2.1.8",
+ "spatialdata": "0.5.0",
+ "spatialdata_io": "0.3.0"
+ },
+ "Workflow": {
+ "nf-core/sopa": "v1.0.0"
+ }
+ },
+ [
+ "pipeline_info",
+ "pipeline_info/nf_core_sopa_software_mqc_versions.yml",
+ "sample_name.explorer",
+ "sample_name.explorer/adata.h5ad",
+ "sample_name.explorer/analysis.zarr.zip",
+ "sample_name.explorer/analysis_summary.html",
+ "sample_name.explorer/cell_feature_matrix.zarr.zip",
+ "sample_name.explorer/cells.zarr.zip",
+ "sample_name.explorer/experiment.xenium",
+ "sample_name.explorer/morphology.ome.tif",
+ "sample_name.explorer/transcripts.zarr.zip",
+ "sample_name.zarr",
+ "sample_name.zarr/.zattrs",
+ "sample_name.zarr/.zgroup",
+ "sample_name.zarr/images",
+ "sample_name.zarr/images/.zgroup",
+ "sample_name.zarr/images/he_image",
+ "sample_name.zarr/images/he_image/.zattrs",
+ "sample_name.zarr/images/he_image/.zgroup",
+ "sample_name.zarr/images/he_image/0",
+ "sample_name.zarr/images/he_image/0/.zarray",
+ "sample_name.zarr/images/he_image/0/0",
+ "sample_name.zarr/images/he_image/0/0/0",
+ "sample_name.zarr/images/he_image/0/0/0/0",
+ "sample_name.zarr/images/he_image/0/0/0/1",
+ "sample_name.zarr/images/he_image/0/0/0/2",
+ "sample_name.zarr/images/he_image/0/0/0/3",
+ "sample_name.zarr/images/he_image/0/0/1",
+ "sample_name.zarr/images/he_image/0/0/1/0",
+ "sample_name.zarr/images/he_image/0/0/1/1",
+ "sample_name.zarr/images/he_image/0/0/1/2",
+ "sample_name.zarr/images/he_image/0/0/1/3",
+ "sample_name.zarr/images/he_image/0/0/2",
+ "sample_name.zarr/images/he_image/0/0/2/0",
+ "sample_name.zarr/images/he_image/0/0/2/1",
+ "sample_name.zarr/images/he_image/0/0/2/2",
+ "sample_name.zarr/images/he_image/0/0/2/3",
+ "sample_name.zarr/images/he_image/0/0/3",
+ "sample_name.zarr/images/he_image/0/0/3/0",
+ "sample_name.zarr/images/he_image/0/0/3/1",
+ "sample_name.zarr/images/he_image/0/0/3/2",
+ "sample_name.zarr/images/he_image/0/0/3/3",
+ "sample_name.zarr/images/he_image/1",
+ "sample_name.zarr/images/he_image/1/.zarray",
+ "sample_name.zarr/images/he_image/1/0",
+ "sample_name.zarr/images/he_image/1/0/0",
+ "sample_name.zarr/images/he_image/1/0/0/0",
+ "sample_name.zarr/images/he_image/1/0/0/1",
+ "sample_name.zarr/images/he_image/1/0/1",
+ "sample_name.zarr/images/he_image/1/0/1/0",
+ "sample_name.zarr/images/he_image/1/0/1/1",
+ "sample_name.zarr/images/he_image/2",
+ "sample_name.zarr/images/he_image/2/.zarray",
+ "sample_name.zarr/images/he_image/2/0",
+ "sample_name.zarr/images/he_image/2/0/0",
+ "sample_name.zarr/images/he_image/2/0/0/0",
+ "sample_name.zarr/images/image",
+ "sample_name.zarr/images/image/.zattrs",
+ "sample_name.zarr/images/image/.zgroup",
+ "sample_name.zarr/images/image/0",
+ "sample_name.zarr/images/image/0/.zarray",
+ "sample_name.zarr/images/image/0/0",
+ "sample_name.zarr/images/image/0/0/0",
+ "sample_name.zarr/images/image/0/0/0/0",
+ "sample_name.zarr/images/image/0/0/0/1",
+ "sample_name.zarr/images/image/0/0/1",
+ "sample_name.zarr/images/image/0/0/1/0",
+ "sample_name.zarr/images/image/0/0/1/1",
+ "sample_name.zarr/images/image/0/1",
+ "sample_name.zarr/images/image/0/1/0",
+ "sample_name.zarr/images/image/0/1/0/0",
+ "sample_name.zarr/images/image/0/1/0/1",
+ "sample_name.zarr/images/image/0/1/1",
+ "sample_name.zarr/images/image/0/1/1/0",
+ "sample_name.zarr/images/image/0/1/1/1",
+ "sample_name.zarr/images/image/0/2",
+ "sample_name.zarr/images/image/0/2/0",
+ "sample_name.zarr/images/image/0/2/0/0",
+ "sample_name.zarr/images/image/0/2/0/1",
+ "sample_name.zarr/images/image/0/2/1",
+ "sample_name.zarr/images/image/0/2/1/0",
+ "sample_name.zarr/images/image/0/2/1/1",
+ "sample_name.zarr/images/image/0/3",
+ "sample_name.zarr/images/image/0/3/0",
+ "sample_name.zarr/images/image/0/3/0/0",
+ "sample_name.zarr/images/image/0/3/0/1",
+ "sample_name.zarr/images/image/0/3/1",
+ "sample_name.zarr/images/image/0/3/1/0",
+ "sample_name.zarr/images/image/0/3/1/1",
+ "sample_name.zarr/points",
+ "sample_name.zarr/points/.zgroup",
+ "sample_name.zarr/points/transcripts",
+ "sample_name.zarr/points/transcripts/.zattrs",
+ "sample_name.zarr/points/transcripts/.zgroup",
+ "sample_name.zarr/points/transcripts/points.parquet",
+ "sample_name.zarr/points/transcripts/points.parquet/part.0.parquet",
+ "sample_name.zarr/shapes",
+ "sample_name.zarr/shapes/.zgroup",
+ "sample_name.zarr/shapes/cellpose_boundaries",
+ "sample_name.zarr/shapes/cellpose_boundaries/.zattrs",
+ "sample_name.zarr/shapes/cellpose_boundaries/.zgroup",
+ "sample_name.zarr/shapes/cellpose_boundaries/shapes.parquet",
+ "sample_name.zarr/shapes/cells",
+ "sample_name.zarr/shapes/cells/.zattrs",
+ "sample_name.zarr/shapes/cells/.zgroup",
+ "sample_name.zarr/shapes/cells/shapes.parquet",
+ "sample_name.zarr/shapes/image_patches",
+ "sample_name.zarr/shapes/image_patches/.zattrs",
+ "sample_name.zarr/shapes/image_patches/.zgroup",
+ "sample_name.zarr/shapes/image_patches/shapes.parquet",
+ "sample_name.zarr/tables",
+ "sample_name.zarr/tables/.zgroup",
+ "sample_name.zarr/tables/table",
+ "sample_name.zarr/tables/table/.zattrs",
+ "sample_name.zarr/tables/table/.zgroup",
+ "sample_name.zarr/tables/table/X",
+ "sample_name.zarr/tables/table/X/.zattrs",
+ "sample_name.zarr/tables/table/X/.zgroup",
+ "sample_name.zarr/tables/table/X/data",
+ "sample_name.zarr/tables/table/X/data/.zarray",
+ "sample_name.zarr/tables/table/X/data/0",
+ "sample_name.zarr/tables/table/X/indices",
+ "sample_name.zarr/tables/table/X/indices/.zarray",
+ "sample_name.zarr/tables/table/X/indices/0",
+ "sample_name.zarr/tables/table/X/indptr",
+ "sample_name.zarr/tables/table/X/indptr/.zarray",
+ "sample_name.zarr/tables/table/X/indptr/0",
+ "sample_name.zarr/tables/table/layers",
+ "sample_name.zarr/tables/table/layers/.zattrs",
+ "sample_name.zarr/tables/table/layers/.zgroup",
+ "sample_name.zarr/tables/table/obs",
+ "sample_name.zarr/tables/table/obs/.zattrs",
+ "sample_name.zarr/tables/table/obs/.zgroup",
+ "sample_name.zarr/tables/table/obs/_index",
+ "sample_name.zarr/tables/table/obs/_index/.zarray",
+ "sample_name.zarr/tables/table/obs/_index/.zattrs",
+ "sample_name.zarr/tables/table/obs/_index/0",
+ "sample_name.zarr/tables/table/obs/area",
+ "sample_name.zarr/tables/table/obs/area/.zarray",
+ "sample_name.zarr/tables/table/obs/area/.zattrs",
+ "sample_name.zarr/tables/table/obs/area/0",
+ "sample_name.zarr/tables/table/obs/cell_id",
+ "sample_name.zarr/tables/table/obs/cell_id/.zarray",
+ "sample_name.zarr/tables/table/obs/cell_id/.zattrs",
+ "sample_name.zarr/tables/table/obs/cell_id/0",
+ "sample_name.zarr/tables/table/obs/region",
+ "sample_name.zarr/tables/table/obs/region/.zattrs",
+ "sample_name.zarr/tables/table/obs/region/.zgroup",
+ "sample_name.zarr/tables/table/obs/region/categories",
+ "sample_name.zarr/tables/table/obs/region/categories/.zarray",
+ "sample_name.zarr/tables/table/obs/region/categories/.zattrs",
+ "sample_name.zarr/tables/table/obs/region/categories/0",
+ "sample_name.zarr/tables/table/obs/region/codes",
+ "sample_name.zarr/tables/table/obs/region/codes/.zarray",
+ "sample_name.zarr/tables/table/obs/region/codes/.zattrs",
+ "sample_name.zarr/tables/table/obs/region/codes/0",
+ "sample_name.zarr/tables/table/obs/slide",
+ "sample_name.zarr/tables/table/obs/slide/.zattrs",
+ "sample_name.zarr/tables/table/obs/slide/.zgroup",
+ "sample_name.zarr/tables/table/obs/slide/categories",
+ "sample_name.zarr/tables/table/obs/slide/categories/.zarray",
+ "sample_name.zarr/tables/table/obs/slide/categories/.zattrs",
+ "sample_name.zarr/tables/table/obs/slide/categories/0",
+ "sample_name.zarr/tables/table/obs/slide/codes",
+ "sample_name.zarr/tables/table/obs/slide/codes/.zarray",
+ "sample_name.zarr/tables/table/obs/slide/codes/.zattrs",
+ "sample_name.zarr/tables/table/obs/slide/codes/0",
+ "sample_name.zarr/tables/table/obsm",
+ "sample_name.zarr/tables/table/obsm/.zattrs",
+ "sample_name.zarr/tables/table/obsm/.zgroup",
+ "sample_name.zarr/tables/table/obsm/intensities",
+ "sample_name.zarr/tables/table/obsm/intensities/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/.zgroup",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20/0",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3/0",
+ "sample_name.zarr/tables/table/obsm/intensities/CK",
+ "sample_name.zarr/tables/table/obsm/intensities/CK/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/CK/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/CK/0",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI/0",
+ "sample_name.zarr/tables/table/obsm/intensities/_index",
+ "sample_name.zarr/tables/table/obsm/intensities/_index/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/_index/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/_index/0",
+ "sample_name.zarr/tables/table/obsm/spatial",
+ "sample_name.zarr/tables/table/obsm/spatial/.zarray",
+ "sample_name.zarr/tables/table/obsm/spatial/.zattrs",
+ "sample_name.zarr/tables/table/obsm/spatial/0",
+ "sample_name.zarr/tables/table/obsm/spatial/0/0",
+ "sample_name.zarr/tables/table/obsp",
+ "sample_name.zarr/tables/table/obsp/.zattrs",
+ "sample_name.zarr/tables/table/obsp/.zgroup",
+ "sample_name.zarr/tables/table/uns",
+ "sample_name.zarr/tables/table/uns/.zattrs",
+ "sample_name.zarr/tables/table/uns/.zgroup",
+ "sample_name.zarr/tables/table/uns/sopa_attrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/.zattrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/.zgroup",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities/.zarray",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities/.zattrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities/0",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts/.zarray",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts/.zattrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts/0",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/.zgroup",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key/.zarray",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key/0",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region/.zarray",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region/0",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key/.zarray",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key/0",
+ "sample_name.zarr/tables/table/var",
+ "sample_name.zarr/tables/table/var/.zattrs",
+ "sample_name.zarr/tables/table/var/.zgroup",
+ "sample_name.zarr/tables/table/var/_index",
+ "sample_name.zarr/tables/table/var/_index/.zarray",
+ "sample_name.zarr/tables/table/var/_index/.zattrs",
+ "sample_name.zarr/tables/table/var/_index/0",
+ "sample_name.zarr/tables/table/varm",
+ "sample_name.zarr/tables/table/varm/.zattrs",
+ "sample_name.zarr/tables/table/varm/.zgroup",
+ "sample_name.zarr/tables/table/varp",
+ "sample_name.zarr/tables/table/varp/.zattrs",
+ "sample_name.zarr/tables/table/varp/.zgroup",
+ "sample_name.zarr/zmetadata"
+ ],
+ [
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab"
+ ]
+ ],
+ "meta": {
+ "nf-test": "0.9.2",
+ "nextflow": "25.04.6"
+ },
+ "timestamp": "2025-08-01T16:10:22.8482"
+ }
+}
\ No newline at end of file
diff --git a/tests/default.nf.test b/tests/default.nf.test
index 9495f6c..3e17df0 100644
--- a/tests/default.nf.test
+++ b/tests/default.nf.test
@@ -2,6 +2,7 @@ nextflow_pipeline {
name "Test pipeline"
script "../main.nf"
+ profile "test"
tag "pipeline"
test("-profile test") {
diff --git a/tests/default.nf.test.snap b/tests/default.nf.test.snap
new file mode 100644
index 0000000..e8f990b
--- /dev/null
+++ b/tests/default.nf.test.snap
@@ -0,0 +1,534 @@
+{
+ "-profile test": {
+ "content": [
+ {
+ "FLUO_ANNOTATION": {
+ "sopa": "2.1.8"
+ },
+ "PATCH_SEGMENTATION_PROSEG": {
+ "sopa": "2.1.8",
+ "proseg": "3.0.10"
+ },
+ "SCANPY_PREPROCESS": {
+ "sopa": "2.1.8",
+ "scanpy": "1.11.5"
+ },
+ "TO_SPATIALDATA": {
+ "sopa": "2.1.8",
+ "spatialdata": "0.5.0",
+ "spatialdata_io": "0.3.0"
+ },
+ "Workflow": {
+ "nf-core/sopa": "v1.0.0"
+ }
+ },
+ [
+ "pipeline_info",
+ "pipeline_info/nf_core_sopa_software_mqc_versions.yml",
+ "sample_name.explorer",
+ "sample_name.explorer/adata.h5ad",
+ "sample_name.explorer/analysis.zarr.zip",
+ "sample_name.explorer/analysis_summary.html",
+ "sample_name.explorer/cell_feature_matrix.zarr.zip",
+ "sample_name.explorer/cells.zarr.zip",
+ "sample_name.explorer/experiment.xenium",
+ "sample_name.explorer/morphology.ome.tif",
+ "sample_name.explorer/transcripts.zarr.zip",
+ "sample_name.zarr",
+ "sample_name.zarr/.zattrs",
+ "sample_name.zarr/.zgroup",
+ "sample_name.zarr/images",
+ "sample_name.zarr/images/.zgroup",
+ "sample_name.zarr/images/he_image",
+ "sample_name.zarr/images/he_image/.zattrs",
+ "sample_name.zarr/images/he_image/.zgroup",
+ "sample_name.zarr/images/he_image/0",
+ "sample_name.zarr/images/he_image/0/.zarray",
+ "sample_name.zarr/images/he_image/0/0",
+ "sample_name.zarr/images/he_image/0/0/0",
+ "sample_name.zarr/images/he_image/0/0/0/0",
+ "sample_name.zarr/images/he_image/0/0/0/1",
+ "sample_name.zarr/images/he_image/0/0/0/2",
+ "sample_name.zarr/images/he_image/0/0/0/3",
+ "sample_name.zarr/images/he_image/0/0/1",
+ "sample_name.zarr/images/he_image/0/0/1/0",
+ "sample_name.zarr/images/he_image/0/0/1/1",
+ "sample_name.zarr/images/he_image/0/0/1/2",
+ "sample_name.zarr/images/he_image/0/0/1/3",
+ "sample_name.zarr/images/he_image/0/0/2",
+ "sample_name.zarr/images/he_image/0/0/2/0",
+ "sample_name.zarr/images/he_image/0/0/2/1",
+ "sample_name.zarr/images/he_image/0/0/2/2",
+ "sample_name.zarr/images/he_image/0/0/2/3",
+ "sample_name.zarr/images/he_image/0/0/3",
+ "sample_name.zarr/images/he_image/0/0/3/0",
+ "sample_name.zarr/images/he_image/0/0/3/1",
+ "sample_name.zarr/images/he_image/0/0/3/2",
+ "sample_name.zarr/images/he_image/0/0/3/3",
+ "sample_name.zarr/images/he_image/1",
+ "sample_name.zarr/images/he_image/1/.zarray",
+ "sample_name.zarr/images/he_image/1/0",
+ "sample_name.zarr/images/he_image/1/0/0",
+ "sample_name.zarr/images/he_image/1/0/0/0",
+ "sample_name.zarr/images/he_image/1/0/0/1",
+ "sample_name.zarr/images/he_image/1/0/1",
+ "sample_name.zarr/images/he_image/1/0/1/0",
+ "sample_name.zarr/images/he_image/1/0/1/1",
+ "sample_name.zarr/images/he_image/2",
+ "sample_name.zarr/images/he_image/2/.zarray",
+ "sample_name.zarr/images/he_image/2/0",
+ "sample_name.zarr/images/he_image/2/0/0",
+ "sample_name.zarr/images/he_image/2/0/0/0",
+ "sample_name.zarr/images/image",
+ "sample_name.zarr/images/image/.zattrs",
+ "sample_name.zarr/images/image/.zgroup",
+ "sample_name.zarr/images/image/0",
+ "sample_name.zarr/images/image/0/.zarray",
+ "sample_name.zarr/images/image/0/0",
+ "sample_name.zarr/images/image/0/0/0",
+ "sample_name.zarr/images/image/0/0/0/0",
+ "sample_name.zarr/images/image/0/0/0/1",
+ "sample_name.zarr/images/image/0/0/1",
+ "sample_name.zarr/images/image/0/0/1/0",
+ "sample_name.zarr/images/image/0/0/1/1",
+ "sample_name.zarr/images/image/0/1",
+ "sample_name.zarr/images/image/0/1/0",
+ "sample_name.zarr/images/image/0/1/0/0",
+ "sample_name.zarr/images/image/0/1/0/1",
+ "sample_name.zarr/images/image/0/1/1",
+ "sample_name.zarr/images/image/0/1/1/0",
+ "sample_name.zarr/images/image/0/1/1/1",
+ "sample_name.zarr/images/image/0/2",
+ "sample_name.zarr/images/image/0/2/0",
+ "sample_name.zarr/images/image/0/2/0/0",
+ "sample_name.zarr/images/image/0/2/0/1",
+ "sample_name.zarr/images/image/0/2/1",
+ "sample_name.zarr/images/image/0/2/1/0",
+ "sample_name.zarr/images/image/0/2/1/1",
+ "sample_name.zarr/images/image/0/3",
+ "sample_name.zarr/images/image/0/3/0",
+ "sample_name.zarr/images/image/0/3/0/0",
+ "sample_name.zarr/images/image/0/3/0/1",
+ "sample_name.zarr/images/image/0/3/1",
+ "sample_name.zarr/images/image/0/3/1/0",
+ "sample_name.zarr/images/image/0/3/1/1",
+ "sample_name.zarr/points",
+ "sample_name.zarr/points/.zgroup",
+ "sample_name.zarr/points/transcripts",
+ "sample_name.zarr/points/transcripts/.zattrs",
+ "sample_name.zarr/points/transcripts/.zgroup",
+ "sample_name.zarr/points/transcripts/points.parquet",
+ "sample_name.zarr/points/transcripts/points.parquet/part.0.parquet",
+ "sample_name.zarr/shapes",
+ "sample_name.zarr/shapes/.zgroup",
+ "sample_name.zarr/shapes/cells",
+ "sample_name.zarr/shapes/cells/.zattrs",
+ "sample_name.zarr/shapes/cells/.zgroup",
+ "sample_name.zarr/shapes/cells/shapes.parquet",
+ "sample_name.zarr/shapes/proseg_boundaries",
+ "sample_name.zarr/shapes/proseg_boundaries/.zattrs",
+ "sample_name.zarr/shapes/proseg_boundaries/.zgroup",
+ "sample_name.zarr/shapes/proseg_boundaries/shapes.parquet",
+ "sample_name.zarr/shapes/transcripts_patches",
+ "sample_name.zarr/shapes/transcripts_patches/.zattrs",
+ "sample_name.zarr/shapes/transcripts_patches/.zgroup",
+ "sample_name.zarr/shapes/transcripts_patches/shapes.parquet",
+ "sample_name.zarr/tables",
+ "sample_name.zarr/tables/.zgroup",
+ "sample_name.zarr/tables/table",
+ "sample_name.zarr/tables/table/.zattrs",
+ "sample_name.zarr/tables/table/.zgroup",
+ "sample_name.zarr/tables/table/X",
+ "sample_name.zarr/tables/table/X/.zattrs",
+ "sample_name.zarr/tables/table/X/.zgroup",
+ "sample_name.zarr/tables/table/X/data",
+ "sample_name.zarr/tables/table/X/data/.zarray",
+ "sample_name.zarr/tables/table/X/data/0",
+ "sample_name.zarr/tables/table/X/indices",
+ "sample_name.zarr/tables/table/X/indices/.zarray",
+ "sample_name.zarr/tables/table/X/indices/0",
+ "sample_name.zarr/tables/table/X/indptr",
+ "sample_name.zarr/tables/table/X/indptr/.zarray",
+ "sample_name.zarr/tables/table/X/indptr/0",
+ "sample_name.zarr/tables/table/layers",
+ "sample_name.zarr/tables/table/layers/.zattrs",
+ "sample_name.zarr/tables/table/layers/.zgroup",
+ "sample_name.zarr/tables/table/layers/counts",
+ "sample_name.zarr/tables/table/layers/counts/.zattrs",
+ "sample_name.zarr/tables/table/layers/counts/.zgroup",
+ "sample_name.zarr/tables/table/layers/counts/data",
+ "sample_name.zarr/tables/table/layers/counts/data/.zarray",
+ "sample_name.zarr/tables/table/layers/counts/data/0",
+ "sample_name.zarr/tables/table/layers/counts/indices",
+ "sample_name.zarr/tables/table/layers/counts/indices/.zarray",
+ "sample_name.zarr/tables/table/layers/counts/indices/0",
+ "sample_name.zarr/tables/table/layers/counts/indptr",
+ "sample_name.zarr/tables/table/layers/counts/indptr/.zarray",
+ "sample_name.zarr/tables/table/layers/counts/indptr/0",
+ "sample_name.zarr/tables/table/obs",
+ "sample_name.zarr/tables/table/obs/.zattrs",
+ "sample_name.zarr/tables/table/obs/.zgroup",
+ "sample_name.zarr/tables/table/obs/_index",
+ "sample_name.zarr/tables/table/obs/_index/.zarray",
+ "sample_name.zarr/tables/table/obs/_index/.zattrs",
+ "sample_name.zarr/tables/table/obs/_index/0",
+ "sample_name.zarr/tables/table/obs/area",
+ "sample_name.zarr/tables/table/obs/area/.zarray",
+ "sample_name.zarr/tables/table/obs/area/.zattrs",
+ "sample_name.zarr/tables/table/obs/area/0",
+ "sample_name.zarr/tables/table/obs/cell",
+ "sample_name.zarr/tables/table/obs/cell/.zarray",
+ "sample_name.zarr/tables/table/obs/cell/.zattrs",
+ "sample_name.zarr/tables/table/obs/cell/0",
+ "sample_name.zarr/tables/table/obs/cell_id",
+ "sample_name.zarr/tables/table/obs/cell_id/.zarray",
+ "sample_name.zarr/tables/table/obs/cell_id/.zattrs",
+ "sample_name.zarr/tables/table/obs/cell_id/0",
+ "sample_name.zarr/tables/table/obs/cell_type",
+ "sample_name.zarr/tables/table/obs/cell_type/.zarray",
+ "sample_name.zarr/tables/table/obs/cell_type/.zattrs",
+ "sample_name.zarr/tables/table/obs/cell_type/0",
+ "sample_name.zarr/tables/table/obs/centroid_x",
+ "sample_name.zarr/tables/table/obs/centroid_x/.zarray",
+ "sample_name.zarr/tables/table/obs/centroid_x/.zattrs",
+ "sample_name.zarr/tables/table/obs/centroid_x/0",
+ "sample_name.zarr/tables/table/obs/centroid_y",
+ "sample_name.zarr/tables/table/obs/centroid_y/.zarray",
+ "sample_name.zarr/tables/table/obs/centroid_y/.zattrs",
+ "sample_name.zarr/tables/table/obs/centroid_y/0",
+ "sample_name.zarr/tables/table/obs/centroid_z",
+ "sample_name.zarr/tables/table/obs/centroid_z/.zarray",
+ "sample_name.zarr/tables/table/obs/centroid_z/.zattrs",
+ "sample_name.zarr/tables/table/obs/centroid_z/0",
+ "sample_name.zarr/tables/table/obs/component",
+ "sample_name.zarr/tables/table/obs/component/.zarray",
+ "sample_name.zarr/tables/table/obs/component/.zattrs",
+ "sample_name.zarr/tables/table/obs/component/0",
+ "sample_name.zarr/tables/table/obs/leiden",
+ "sample_name.zarr/tables/table/obs/leiden/.zattrs",
+ "sample_name.zarr/tables/table/obs/leiden/.zgroup",
+ "sample_name.zarr/tables/table/obs/leiden/categories",
+ "sample_name.zarr/tables/table/obs/leiden/categories/.zarray",
+ "sample_name.zarr/tables/table/obs/leiden/categories/.zattrs",
+ "sample_name.zarr/tables/table/obs/leiden/categories/0",
+ "sample_name.zarr/tables/table/obs/leiden/codes",
+ "sample_name.zarr/tables/table/obs/leiden/codes/.zarray",
+ "sample_name.zarr/tables/table/obs/leiden/codes/.zattrs",
+ "sample_name.zarr/tables/table/obs/leiden/codes/0",
+ "sample_name.zarr/tables/table/obs/original_cell_id",
+ "sample_name.zarr/tables/table/obs/original_cell_id/.zarray",
+ "sample_name.zarr/tables/table/obs/original_cell_id/.zattrs",
+ "sample_name.zarr/tables/table/obs/original_cell_id/0",
+ "sample_name.zarr/tables/table/obs/region",
+ "sample_name.zarr/tables/table/obs/region/.zattrs",
+ "sample_name.zarr/tables/table/obs/region/.zgroup",
+ "sample_name.zarr/tables/table/obs/region/categories",
+ "sample_name.zarr/tables/table/obs/region/categories/.zarray",
+ "sample_name.zarr/tables/table/obs/region/categories/.zattrs",
+ "sample_name.zarr/tables/table/obs/region/categories/0",
+ "sample_name.zarr/tables/table/obs/region/codes",
+ "sample_name.zarr/tables/table/obs/region/codes/.zarray",
+ "sample_name.zarr/tables/table/obs/region/codes/.zattrs",
+ "sample_name.zarr/tables/table/obs/region/codes/0",
+ "sample_name.zarr/tables/table/obs/scale",
+ "sample_name.zarr/tables/table/obs/scale/.zarray",
+ "sample_name.zarr/tables/table/obs/scale/.zattrs",
+ "sample_name.zarr/tables/table/obs/scale/0",
+ "sample_name.zarr/tables/table/obs/slide",
+ "sample_name.zarr/tables/table/obs/slide/.zattrs",
+ "sample_name.zarr/tables/table/obs/slide/.zgroup",
+ "sample_name.zarr/tables/table/obs/slide/categories",
+ "sample_name.zarr/tables/table/obs/slide/categories/.zarray",
+ "sample_name.zarr/tables/table/obs/slide/categories/.zattrs",
+ "sample_name.zarr/tables/table/obs/slide/categories/0",
+ "sample_name.zarr/tables/table/obs/slide/codes",
+ "sample_name.zarr/tables/table/obs/slide/codes/.zarray",
+ "sample_name.zarr/tables/table/obs/slide/codes/.zattrs",
+ "sample_name.zarr/tables/table/obs/slide/codes/0",
+ "sample_name.zarr/tables/table/obs/surface_area",
+ "sample_name.zarr/tables/table/obs/surface_area/.zarray",
+ "sample_name.zarr/tables/table/obs/surface_area/.zattrs",
+ "sample_name.zarr/tables/table/obs/surface_area/0",
+ "sample_name.zarr/tables/table/obs/volume",
+ "sample_name.zarr/tables/table/obs/volume/.zarray",
+ "sample_name.zarr/tables/table/obs/volume/.zattrs",
+ "sample_name.zarr/tables/table/obs/volume/0",
+ "sample_name.zarr/tables/table/obsm",
+ "sample_name.zarr/tables/table/obsm/.zattrs",
+ "sample_name.zarr/tables/table/obsm/.zgroup",
+ "sample_name.zarr/tables/table/obsm/X_pca",
+ "sample_name.zarr/tables/table/obsm/X_pca/.zarray",
+ "sample_name.zarr/tables/table/obsm/X_pca/.zattrs",
+ "sample_name.zarr/tables/table/obsm/X_pca/0",
+ "sample_name.zarr/tables/table/obsm/X_pca/0/0",
+ "sample_name.zarr/tables/table/obsm/X_umap",
+ "sample_name.zarr/tables/table/obsm/X_umap/.zarray",
+ "sample_name.zarr/tables/table/obsm/X_umap/.zattrs",
+ "sample_name.zarr/tables/table/obsm/X_umap/0",
+ "sample_name.zarr/tables/table/obsm/X_umap/0/0",
+ "sample_name.zarr/tables/table/obsm/intensities",
+ "sample_name.zarr/tables/table/obsm/intensities/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/.zgroup",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/CD20/0",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/CD3/0",
+ "sample_name.zarr/tables/table/obsm/intensities/CK",
+ "sample_name.zarr/tables/table/obsm/intensities/CK/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/CK/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/CK/0",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/DAPI/0",
+ "sample_name.zarr/tables/table/obsm/intensities/_index",
+ "sample_name.zarr/tables/table/obsm/intensities/_index/.zarray",
+ "sample_name.zarr/tables/table/obsm/intensities/_index/.zattrs",
+ "sample_name.zarr/tables/table/obsm/intensities/_index/0",
+ "sample_name.zarr/tables/table/obsm/spatial",
+ "sample_name.zarr/tables/table/obsm/spatial/.zarray",
+ "sample_name.zarr/tables/table/obsm/spatial/.zattrs",
+ "sample_name.zarr/tables/table/obsm/spatial/0",
+ "sample_name.zarr/tables/table/obsm/spatial/0/0",
+ "sample_name.zarr/tables/table/obsm/z_scores",
+ "sample_name.zarr/tables/table/obsm/z_scores/.zattrs",
+ "sample_name.zarr/tables/table/obsm/z_scores/.zgroup",
+ "sample_name.zarr/tables/table/obsm/z_scores/CD20",
+ "sample_name.zarr/tables/table/obsm/z_scores/CD20/.zarray",
+ "sample_name.zarr/tables/table/obsm/z_scores/CD20/.zattrs",
+ "sample_name.zarr/tables/table/obsm/z_scores/CD20/0",
+ "sample_name.zarr/tables/table/obsm/z_scores/CD3",
+ "sample_name.zarr/tables/table/obsm/z_scores/CD3/.zarray",
+ "sample_name.zarr/tables/table/obsm/z_scores/CD3/.zattrs",
+ "sample_name.zarr/tables/table/obsm/z_scores/CD3/0",
+ "sample_name.zarr/tables/table/obsm/z_scores/CK",
+ "sample_name.zarr/tables/table/obsm/z_scores/CK/.zarray",
+ "sample_name.zarr/tables/table/obsm/z_scores/CK/.zattrs",
+ "sample_name.zarr/tables/table/obsm/z_scores/CK/0",
+ "sample_name.zarr/tables/table/obsm/z_scores/DAPI",
+ "sample_name.zarr/tables/table/obsm/z_scores/DAPI/.zarray",
+ "sample_name.zarr/tables/table/obsm/z_scores/DAPI/.zattrs",
+ "sample_name.zarr/tables/table/obsm/z_scores/DAPI/0",
+ "sample_name.zarr/tables/table/obsm/z_scores/_index",
+ "sample_name.zarr/tables/table/obsm/z_scores/_index/.zarray",
+ "sample_name.zarr/tables/table/obsm/z_scores/_index/.zattrs",
+ "sample_name.zarr/tables/table/obsm/z_scores/_index/0",
+ "sample_name.zarr/tables/table/obsp",
+ "sample_name.zarr/tables/table/obsp/.zattrs",
+ "sample_name.zarr/tables/table/obsp/.zgroup",
+ "sample_name.zarr/tables/table/obsp/connectivities",
+ "sample_name.zarr/tables/table/obsp/connectivities/.zattrs",
+ "sample_name.zarr/tables/table/obsp/connectivities/.zgroup",
+ "sample_name.zarr/tables/table/obsp/connectivities/data",
+ "sample_name.zarr/tables/table/obsp/connectivities/data/.zarray",
+ "sample_name.zarr/tables/table/obsp/connectivities/data/0",
+ "sample_name.zarr/tables/table/obsp/connectivities/indices",
+ "sample_name.zarr/tables/table/obsp/connectivities/indices/.zarray",
+ "sample_name.zarr/tables/table/obsp/connectivities/indices/0",
+ "sample_name.zarr/tables/table/obsp/connectivities/indptr",
+ "sample_name.zarr/tables/table/obsp/connectivities/indptr/.zarray",
+ "sample_name.zarr/tables/table/obsp/connectivities/indptr/0",
+ "sample_name.zarr/tables/table/obsp/distances",
+ "sample_name.zarr/tables/table/obsp/distances/.zattrs",
+ "sample_name.zarr/tables/table/obsp/distances/.zgroup",
+ "sample_name.zarr/tables/table/obsp/distances/data",
+ "sample_name.zarr/tables/table/obsp/distances/data/.zarray",
+ "sample_name.zarr/tables/table/obsp/distances/data/0",
+ "sample_name.zarr/tables/table/obsp/distances/indices",
+ "sample_name.zarr/tables/table/obsp/distances/indices/.zarray",
+ "sample_name.zarr/tables/table/obsp/distances/indices/0",
+ "sample_name.zarr/tables/table/obsp/distances/indptr",
+ "sample_name.zarr/tables/table/obsp/distances/indptr/.zarray",
+ "sample_name.zarr/tables/table/obsp/distances/indptr/0",
+ "sample_name.zarr/tables/table/uns",
+ "sample_name.zarr/tables/table/uns/.zattrs",
+ "sample_name.zarr/tables/table/uns/.zgroup",
+ "sample_name.zarr/tables/table/uns/leiden",
+ "sample_name.zarr/tables/table/uns/leiden/.zattrs",
+ "sample_name.zarr/tables/table/uns/leiden/.zgroup",
+ "sample_name.zarr/tables/table/uns/leiden/params",
+ "sample_name.zarr/tables/table/uns/leiden/params/.zattrs",
+ "sample_name.zarr/tables/table/uns/leiden/params/.zgroup",
+ "sample_name.zarr/tables/table/uns/leiden/params/n_iterations",
+ "sample_name.zarr/tables/table/uns/leiden/params/n_iterations/.zarray",
+ "sample_name.zarr/tables/table/uns/leiden/params/n_iterations/.zattrs",
+ "sample_name.zarr/tables/table/uns/leiden/params/n_iterations/0",
+ "sample_name.zarr/tables/table/uns/leiden/params/random_state",
+ "sample_name.zarr/tables/table/uns/leiden/params/random_state/.zarray",
+ "sample_name.zarr/tables/table/uns/leiden/params/random_state/.zattrs",
+ "sample_name.zarr/tables/table/uns/leiden/params/random_state/0",
+ "sample_name.zarr/tables/table/uns/leiden/params/resolution",
+ "sample_name.zarr/tables/table/uns/leiden/params/resolution/.zarray",
+ "sample_name.zarr/tables/table/uns/leiden/params/resolution/.zattrs",
+ "sample_name.zarr/tables/table/uns/leiden/params/resolution/0",
+ "sample_name.zarr/tables/table/uns/log1p",
+ "sample_name.zarr/tables/table/uns/log1p/.zattrs",
+ "sample_name.zarr/tables/table/uns/log1p/.zgroup",
+ "sample_name.zarr/tables/table/uns/neighbors",
+ "sample_name.zarr/tables/table/uns/neighbors/.zattrs",
+ "sample_name.zarr/tables/table/uns/neighbors/.zgroup",
+ "sample_name.zarr/tables/table/uns/neighbors/connectivities_key",
+ "sample_name.zarr/tables/table/uns/neighbors/connectivities_key/.zarray",
+ "sample_name.zarr/tables/table/uns/neighbors/connectivities_key/.zattrs",
+ "sample_name.zarr/tables/table/uns/neighbors/connectivities_key/0",
+ "sample_name.zarr/tables/table/uns/neighbors/distances_key",
+ "sample_name.zarr/tables/table/uns/neighbors/distances_key/.zarray",
+ "sample_name.zarr/tables/table/uns/neighbors/distances_key/.zattrs",
+ "sample_name.zarr/tables/table/uns/neighbors/distances_key/0",
+ "sample_name.zarr/tables/table/uns/neighbors/params",
+ "sample_name.zarr/tables/table/uns/neighbors/params/.zattrs",
+ "sample_name.zarr/tables/table/uns/neighbors/params/.zgroup",
+ "sample_name.zarr/tables/table/uns/neighbors/params/method",
+ "sample_name.zarr/tables/table/uns/neighbors/params/method/.zarray",
+ "sample_name.zarr/tables/table/uns/neighbors/params/method/.zattrs",
+ "sample_name.zarr/tables/table/uns/neighbors/params/method/0",
+ "sample_name.zarr/tables/table/uns/neighbors/params/metric",
+ "sample_name.zarr/tables/table/uns/neighbors/params/metric/.zarray",
+ "sample_name.zarr/tables/table/uns/neighbors/params/metric/.zattrs",
+ "sample_name.zarr/tables/table/uns/neighbors/params/metric/0",
+ "sample_name.zarr/tables/table/uns/neighbors/params/n_neighbors",
+ "sample_name.zarr/tables/table/uns/neighbors/params/n_neighbors/.zarray",
+ "sample_name.zarr/tables/table/uns/neighbors/params/n_neighbors/.zattrs",
+ "sample_name.zarr/tables/table/uns/neighbors/params/n_neighbors/0",
+ "sample_name.zarr/tables/table/uns/neighbors/params/random_state",
+ "sample_name.zarr/tables/table/uns/neighbors/params/random_state/.zarray",
+ "sample_name.zarr/tables/table/uns/neighbors/params/random_state/.zattrs",
+ "sample_name.zarr/tables/table/uns/neighbors/params/random_state/0",
+ "sample_name.zarr/tables/table/uns/pca",
+ "sample_name.zarr/tables/table/uns/pca/.zattrs",
+ "sample_name.zarr/tables/table/uns/pca/.zgroup",
+ "sample_name.zarr/tables/table/uns/pca/params",
+ "sample_name.zarr/tables/table/uns/pca/params/.zattrs",
+ "sample_name.zarr/tables/table/uns/pca/params/.zgroup",
+ "sample_name.zarr/tables/table/uns/pca/params/use_highly_variable",
+ "sample_name.zarr/tables/table/uns/pca/params/use_highly_variable/.zarray",
+ "sample_name.zarr/tables/table/uns/pca/params/use_highly_variable/.zattrs",
+ "sample_name.zarr/tables/table/uns/pca/params/use_highly_variable/0",
+ "sample_name.zarr/tables/table/uns/pca/params/zero_center",
+ "sample_name.zarr/tables/table/uns/pca/params/zero_center/.zarray",
+ "sample_name.zarr/tables/table/uns/pca/params/zero_center/.zattrs",
+ "sample_name.zarr/tables/table/uns/pca/params/zero_center/0",
+ "sample_name.zarr/tables/table/uns/pca/variance",
+ "sample_name.zarr/tables/table/uns/pca/variance/.zarray",
+ "sample_name.zarr/tables/table/uns/pca/variance/.zattrs",
+ "sample_name.zarr/tables/table/uns/pca/variance/0",
+ "sample_name.zarr/tables/table/uns/pca/variance_ratio",
+ "sample_name.zarr/tables/table/uns/pca/variance_ratio/.zarray",
+ "sample_name.zarr/tables/table/uns/pca/variance_ratio/.zattrs",
+ "sample_name.zarr/tables/table/uns/pca/variance_ratio/0",
+ "sample_name.zarr/tables/table/uns/proseg_run",
+ "sample_name.zarr/tables/table/uns/proseg_run/.zattrs",
+ "sample_name.zarr/tables/table/uns/proseg_run/.zgroup",
+ "sample_name.zarr/tables/table/uns/proseg_run/args",
+ "sample_name.zarr/tables/table/uns/proseg_run/args/.zarray",
+ "sample_name.zarr/tables/table/uns/proseg_run/args/.zattrs",
+ "sample_name.zarr/tables/table/uns/proseg_run/args/0",
+ "sample_name.zarr/tables/table/uns/proseg_run/duration",
+ "sample_name.zarr/tables/table/uns/proseg_run/duration/.zarray",
+ "sample_name.zarr/tables/table/uns/proseg_run/duration/.zattrs",
+ "sample_name.zarr/tables/table/uns/proseg_run/duration/0",
+ "sample_name.zarr/tables/table/uns/proseg_run/version",
+ "sample_name.zarr/tables/table/uns/proseg_run/version/.zarray",
+ "sample_name.zarr/tables/table/uns/proseg_run/version/.zattrs",
+ "sample_name.zarr/tables/table/uns/proseg_run/version/0",
+ "sample_name.zarr/tables/table/uns/sopa_attrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/.zattrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/.zgroup",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/cell_types",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/cell_types/.zarray",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/cell_types/.zattrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/cell_types/0",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities/.zarray",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities/.zattrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/intensities/0",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts/.zarray",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts/.zattrs",
+ "sample_name.zarr/tables/table/uns/sopa_attrs/transcripts/0",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/.zgroup",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key/.zarray",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/instance_key/0",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region/.zarray",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region/0",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key/.zarray",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key/.zattrs",
+ "sample_name.zarr/tables/table/uns/spatialdata_attrs/region_key/0",
+ "sample_name.zarr/tables/table/uns/umap",
+ "sample_name.zarr/tables/table/uns/umap/.zattrs",
+ "sample_name.zarr/tables/table/uns/umap/.zgroup",
+ "sample_name.zarr/tables/table/uns/umap/params",
+ "sample_name.zarr/tables/table/uns/umap/params/.zattrs",
+ "sample_name.zarr/tables/table/uns/umap/params/.zgroup",
+ "sample_name.zarr/tables/table/uns/umap/params/a",
+ "sample_name.zarr/tables/table/uns/umap/params/a/.zarray",
+ "sample_name.zarr/tables/table/uns/umap/params/a/.zattrs",
+ "sample_name.zarr/tables/table/uns/umap/params/a/0",
+ "sample_name.zarr/tables/table/uns/umap/params/b",
+ "sample_name.zarr/tables/table/uns/umap/params/b/.zarray",
+ "sample_name.zarr/tables/table/uns/umap/params/b/.zattrs",
+ "sample_name.zarr/tables/table/uns/umap/params/b/0",
+ "sample_name.zarr/tables/table/var",
+ "sample_name.zarr/tables/table/var/.zattrs",
+ "sample_name.zarr/tables/table/var/.zgroup",
+ "sample_name.zarr/tables/table/var/_index",
+ "sample_name.zarr/tables/table/var/_index/.zarray",
+ "sample_name.zarr/tables/table/var/_index/.zattrs",
+ "sample_name.zarr/tables/table/var/_index/0",
+ "sample_name.zarr/tables/table/var/gene",
+ "sample_name.zarr/tables/table/var/gene/.zarray",
+ "sample_name.zarr/tables/table/var/gene/.zattrs",
+ "sample_name.zarr/tables/table/var/gene/0",
+ "sample_name.zarr/tables/table/var/lambda_bg_0",
+ "sample_name.zarr/tables/table/var/lambda_bg_0/.zarray",
+ "sample_name.zarr/tables/table/var/lambda_bg_0/.zattrs",
+ "sample_name.zarr/tables/table/var/lambda_bg_0/0",
+ "sample_name.zarr/tables/table/var/total_count",
+ "sample_name.zarr/tables/table/var/total_count/.zarray",
+ "sample_name.zarr/tables/table/var/total_count/.zattrs",
+ "sample_name.zarr/tables/table/var/total_count/0",
+ "sample_name.zarr/tables/table/varm",
+ "sample_name.zarr/tables/table/varm/.zattrs",
+ "sample_name.zarr/tables/table/varm/.zgroup",
+ "sample_name.zarr/tables/table/varm/PCs",
+ "sample_name.zarr/tables/table/varm/PCs/.zarray",
+ "sample_name.zarr/tables/table/varm/PCs/.zattrs",
+ "sample_name.zarr/tables/table/varm/PCs/0",
+ "sample_name.zarr/tables/table/varm/PCs/0/0",
+ "sample_name.zarr/tables/table/varp",
+ "sample_name.zarr/tables/table/varp/.zattrs",
+ "sample_name.zarr/tables/table/varp/.zgroup",
+ "sample_name.zarr/zmetadata"
+ ],
+ [
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab",
+ ".zgroup:md5,e20297935e73dd0154104d4ea53040ab"
+ ]
+ ],
+ "meta": {
+ "nf-test": "0.9.2",
+ "nextflow": "25.04.6"
+ },
+ "timestamp": "2025-10-15T11:11:22.043613"
+ }
+}
\ No newline at end of file
diff --git a/tests/nextflow.config b/tests/nextflow.config
index acd0dbf..9c78692 100644
--- a/tests/nextflow.config
+++ b/tests/nextflow.config
@@ -4,11 +4,9 @@
========================================================================================
*/
-// TODO nf-core: Specify any additional parameters here
-// Or any resources requirements
params {
modules_testdata_base_path = 'https://raw.githubusercontent.com/nf-core/test-datasets/modules/data/'
pipelines_testdata_base_path = 'https://raw.githubusercontent.com/nf-core/test-datasets/refs/heads/sopa'
}
-aws.client.anonymous = true // fixes S3 access issues on self-hosted runners
+aws.client.anonymous = true
diff --git a/tests/samplesheet.csv b/tests/samplesheet.csv
new file mode 100644
index 0000000..5f0dd9c
--- /dev/null
+++ b/tests/samplesheet.csv
@@ -0,0 +1,2 @@
+sample,data_path
+sample_name,https://github.com/nf-core/sopa/blob/dev/tests/samplesheet.csv
diff --git a/tests/samplesheet_visium_hd.csv b/tests/samplesheet_visium_hd.csv
new file mode 100644
index 0000000..00acae2
--- /dev/null
+++ b/tests/samplesheet_visium_hd.csv
@@ -0,0 +1,2 @@
+sample,fastq_dir,image,cytaimage,slide,area
+Visium_HD_Human_Lung_Cancer_Fixed_Frozen,Visium_HD_Human_Lung_Cancer_Fixed_Frozen/Visium_HD_Human_Lung_Cancer_Fixed_Frozen_fastqs,Visium_HD_Human_Lung_Cancer_Fixed_Frozen/Visium_HD_Human_Lung_Cancer_Fixed_Frozen_tissue_image.btf,Visium_HD_Human_Lung_Cancer_Fixed_Frozen/Visium_HD_Human_Lung_Cancer_Fixed_Frozen_image.tif,H1-TY834G7,D1
diff --git a/tower.yml b/tower.yml
deleted file mode 100644
index c61323c..0000000
--- a/tower.yml
+++ /dev/null
@@ -1,3 +0,0 @@
-reports:
- samplesheet.csv:
- display: "Auto-created samplesheet with collated metadata and FASTQ paths"
diff --git a/workflows/sopa.nf b/workflows/sopa.nf
index 1adbfff..33c20e4 100644
--- a/workflows/sopa.nf
+++ b/workflows/sopa.nf
@@ -3,10 +3,32 @@
IMPORT MODULES / SUBWORKFLOWS / FUNCTIONS
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
*/
-include { paramsSummaryMap } from 'plugin/nf-schema'
+include { paramsSummaryMap } from 'plugin/nf-schema'
include { softwareVersionsToYAML } from '../subworkflows/nf-core/utils_nfcore_pipeline'
include { methodsDescriptionText } from '../subworkflows/local/utils_nfcore_sopa_pipeline'
+include { TO_SPATIALDATA } from '../modules/local/to_spatialdata'
+include { MAKE_IMAGE_PATCHES } from '../modules/local/make_image_patches'
+include { MAKE_TRANSCRIPT_PATCHES } from '../modules/local/make_transcript_patches'
+include { TISSUE_SEGMENTATION } from '../modules/local/tissue_segmentation'
+include { PATCH_SEGMENTATION_BAYSOR } from '../modules/local/patch_segmentation_baysor'
+include { PATCH_SEGMENTATION_COMSEG } from '../modules/local/patch_segmentation_comseg'
+include { PATCH_SEGMENTATION_CELLPOSE } from '../modules/local/patch_segmentation_cellpose'
+include { PATCH_SEGMENTATION_STARDIST } from '../modules/local/patch_segmentation_stardist'
+include { PATCH_SEGMENTATION_PROSEG } from '../modules/local/patch_segmentation_proseg'
+include { RESOLVE_BAYSOR } from '../modules/local/resolve_baysor'
+include { RESOLVE_COMSEG } from '../modules/local/resolve_comseg'
+include { RESOLVE_CELLPOSE } from '../modules/local/resolve_cellpose'
+include { RESOLVE_STARDIST } from '../modules/local/resolve_stardist'
+include { AGGREGATE } from '../modules/local/aggregate'
+include { EXPLORER } from '../modules/local/explorer'
+include { EXPLORER_RAW } from '../modules/local/explorer_raw'
+include { SCANPY_PREPROCESS } from '../modules/local/scanpy_preprocess'
+include { REPORT } from '../modules/local/report'
+include { TANGRAM_ANNOTATION } from '../modules/local/tangram_annotation'
+include { FLUO_ANNOTATION } from '../modules/local/fluo_annotation'
+include { SPACERANGER } from '../subworkflows/local/spaceranger'
+include { ArgsCLI } from '../modules/local/utils'
/*
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
RUN MAIN WORKFLOW
@@ -14,32 +36,271 @@ include { methodsDescriptionText } from '../subworkflows/local/utils_nfcore_sopa
*/
workflow SOPA {
-
take:
ch_samplesheet // channel: samplesheet read in from --input
+
main:
ch_versions = Channel.empty()
+ if (params.read.technology == "visium_hd") {
+ (ch_input_spatialdata, versions) = SPACERANGER(ch_samplesheet)
+ ch_input_spatialdata = ch_input_spatialdata.map { meta, out -> [meta, out[0].toString().replaceFirst(/(.*?outs).*/, '$1'), meta.image] }
+
+ ch_versions = ch_versions.mix(versions)
+ }
+ else {
+ ch_input_spatialdata = ch_samplesheet.map { meta -> [meta, meta.data_dir, []] }
+ }
+
+ (ch_spatialdata, versions) = TO_SPATIALDATA(ch_input_spatialdata)
+ ch_versions = ch_versions.mix(versions)
+
+ ch_explorer_raw = ch_spatialdata.map { meta, sdata_path -> [meta, sdata_path, params.read.technology == "xenium" ? meta.data_dir : []] }
+ EXPLORER_RAW(ch_explorer_raw)
+
+ if (params.segmentation.tissue) {
+ (ch_tissue_seg, _out) = TISSUE_SEGMENTATION(ch_spatialdata, ArgsCLI(params.segmentation.tissue))
+ }
+ else {
+ ch_tissue_seg = ch_spatialdata
+ }
+
+ if (params.segmentation.cellpose) {
+ (ch_image_patches, _out) = MAKE_IMAGE_PATCHES(ch_tissue_seg, ArgsCLI(params.patchify, "pixel"))
+ (ch_resolved, versions) = CELLPOSE(ch_image_patches, params)
+
+ ch_versions = ch_versions.mix(versions)
+ }
+
+ if (params.segmentation.stardist) {
+ (ch_image_patches, _out) = MAKE_IMAGE_PATCHES(ch_tissue_seg, ArgsCLI(params.patchify, "pixel"))
+ (ch_resolved, versions) = STARDIST(ch_image_patches, params)
+
+ ch_versions = ch_versions.mix(versions)
+ }
+
+ if (params.segmentation.baysor) {
+ ch_input_baysor = params.segmentation.cellpose ? ch_resolved : ch_tissue_seg
+
+ ch_transcripts_patches = MAKE_TRANSCRIPT_PATCHES(ch_input_baysor, transcriptPatchesArgs(params, "baysor"))
+ (ch_resolved, versions) = BAYSOR(ch_transcripts_patches, params)
+
+ ch_versions = ch_versions.mix(versions)
+ }
+
+ if (params.segmentation.comseg) {
+ ch_input_comseg = params.segmentation.cellpose ? ch_resolved : ch_tissue_seg
+
+ ch_transcripts_patches = MAKE_TRANSCRIPT_PATCHES(ch_input_comseg, transcriptPatchesArgs(params, "comseg"))
+ (ch_resolved, versions) = COMSEG(ch_transcripts_patches, params)
+
+ ch_versions = ch_versions.mix(versions)
+ }
+
+ if (params.segmentation.proseg) {
+ ch_input_proseg = params.segmentation.cellpose ? ch_resolved : ch_tissue_seg
+
+ ch_proseg_patches = MAKE_TRANSCRIPT_PATCHES(ch_input_proseg, transcriptPatchesArgs(params, "proseg"))
+ (ch_resolved, versions) = PROSEG(ch_proseg_patches, params)
+
+ ch_versions = ch_versions.mix(versions)
+ }
+
+ (ch_aggregated, _out) = AGGREGATE(ch_resolved, ArgsCLI(params.aggregate))
+
+ if (params.annotation && params.annotation.method == "tangram") {
+ sc_reference = file(params.annotation.args.sc_reference_path)
+ params.annotation.args.remove('sc_reference_path')
+
+ (ch_annotated, _out, versions) = TANGRAM_ANNOTATION(ch_aggregated, sc_reference, ArgsCLI(params.annotation.args))
+ ch_versions = ch_versions.mix(versions)
+ }
+ else if (params.annotation && params.annotation.method == "fluorescence") {
+ (ch_annotated, _out, versions) = FLUO_ANNOTATION(ch_aggregated, ArgsCLI(params.annotation.args))
+ ch_versions = ch_versions.mix(versions)
+ }
+ else {
+ ch_annotated = ch_aggregated
+ }
+
+ if (params.scanpy_preprocess) {
+ (ch_preprocessed, _out, versions) = SCANPY_PREPROCESS(ch_annotated, ArgsCLI(params.scanpy_preprocess))
+ ch_versions = ch_versions.mix(versions)
+ }
+ else {
+ ch_preprocessed = ch_annotated
+ }
+
+ EXPLORER(ch_preprocessed, ArgsCLI(params.explorer))
+
+ REPORT(ch_preprocessed)
+
//
// Collate and save software versions
//
- softwareVersionsToYAML(ch_versions)
- .collectFile(
- storeDir: "${params.outdir}/pipeline_info",
- name: 'nf_core_' + 'sopa_software_' + 'versions.yml',
- sort: true,
- newLine: true
- ).set { ch_collated_versions }
-
+ softwareVersionsToYAML(ch_versions).collectFile(
+ storeDir: "${params.outdir}/pipeline_info",
+ name: 'nf_core_sopa_software_mqc_versions.yml',
+ sort: true,
+ newLine: true,
+ )
emit:
- versions = ch_versions // channel: [ path(versions.yml) ]
-
+ versions = ch_versions // channel: [ path(versions.yml) ]
}
/*
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
- THE END
+ SEGMENTATION WORKFLOWS
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
*/
+
+workflow CELLPOSE {
+ take:
+ ch_patches
+ config
+
+ main:
+ ch_versions = Channel.empty()
+
+ cellpose_args = ArgsCLI(config.segmentation.cellpose)
+
+ ch_patches
+ .map { meta, sdata_path, patches_file_image -> [meta, sdata_path, patches_file_image.text.trim().toInteger()] }
+ .flatMap { meta, sdata_path, n_patches -> (0.. [meta, sdata_path, cellpose_args, index, n_patches] } }
+ .set { ch_cellpose }
+
+ ch_segmented = PATCH_SEGMENTATION_CELLPOSE(ch_cellpose).map { meta, sdata_path, _out, n_patches -> [groupKey(meta.sdata_dir, n_patches), [meta, sdata_path]] }.groupTuple().map { it -> it[1][0] }
+
+ (ch_resolved, _out, versions) = RESOLVE_CELLPOSE(ch_segmented)
+
+ ch_versions = ch_versions.mix(versions)
+
+ emit:
+ ch_resolved
+ ch_versions
+}
+
+workflow STARDIST {
+ take:
+ ch_patches
+ config
+
+ main:
+ ch_versions = Channel.empty()
+
+ stardist_args = ArgsCLI(config.segmentation.stardist)
+
+ ch_patches
+ .map { meta, sdata_path, patches_file_image -> [meta, sdata_path, patches_file_image.text.trim().toInteger()] }
+ .flatMap { meta, sdata_path, n_patches -> (0.. [meta, sdata_path, stardist_args, index, n_patches] } }
+ .set { ch_stardist }
+
+ ch_segmented = PATCH_SEGMENTATION_STARDIST(ch_stardist).map { meta, sdata_path, _out, n_patches -> [groupKey(meta.sdata_dir, n_patches), [meta, sdata_path]] }.groupTuple().map { it -> it[1][0] }
+
+ (ch_resolved, _out, versions) = RESOLVE_STARDIST(ch_segmented)
+
+ ch_versions = ch_versions.mix(versions)
+
+ emit:
+ ch_resolved
+ ch_versions
+}
+
+workflow PROSEG {
+ take:
+ ch_patches
+ config
+
+ main:
+ ch_versions = Channel.empty()
+
+ proseg_args = ArgsCLI(config.segmentation.proseg, null, ["command_line_suffix"])
+
+ (ch_segmented, _out, versions) = PATCH_SEGMENTATION_PROSEG(ch_patches, proseg_args)
+
+ ch_versions = ch_versions.mix(versions)
+
+ emit:
+ ch_segmented
+ ch_versions
+}
+
+
+workflow BAYSOR {
+ take:
+ ch_patches
+ config
+
+ main:
+ ch_versions = Channel.empty()
+
+ baysor_args = ArgsCLI(config.segmentation.baysor, null, ["config"])
+
+ ch_patches
+ .map { meta, sdata_path, patches_file_transcripts, _patches -> [meta, sdata_path, patches_file_transcripts.splitText()] }
+ .flatMap { meta, sdata_path, patches_indices -> patches_indices.collect { index -> [meta, sdata_path, baysor_args, index.trim().toInteger(), patches_indices.size] } }
+ .set { ch_baysor }
+
+ ch_segmented = PATCH_SEGMENTATION_BAYSOR(ch_baysor).map { meta, sdata_path, _out, n_patches -> [groupKey(meta.sdata_dir, n_patches), [meta, sdata_path]] }.groupTuple().map { it -> it[1][0] }
+
+ (ch_resolved, _out, versions) = RESOLVE_BAYSOR(ch_segmented, resolveArgs(config))
+
+ ch_versions = ch_versions.mix(versions)
+
+ emit:
+ ch_resolved
+ ch_versions
+}
+
+workflow COMSEG {
+ take:
+ ch_patches
+ config
+
+ main:
+ ch_versions = Channel.empty()
+
+ comseg_args = ArgsCLI(config.segmentation.comseg, null, ["config"])
+
+ ch_patches
+ .map { meta, sdata_path, patches_file_transcripts, _patches -> [meta, sdata_path, patches_file_transcripts.splitText()] }
+ .flatMap { meta, sdata_path, patches_indices -> patches_indices.collect { index -> [meta, sdata_path, comseg_args, index.trim().toInteger(), patches_indices.size] } }
+ .set { ch_comseg }
+
+ ch_segmented = PATCH_SEGMENTATION_COMSEG(ch_comseg).map { meta, sdata_path, _out1, _out2, n_patches -> [groupKey(meta.sdata_dir, n_patches), [meta, sdata_path]] }.groupTuple().map { it -> it[1][0] }
+
+ (ch_resolved, _out, versions) = RESOLVE_COMSEG(ch_segmented, resolveArgs(config))
+
+ ch_versions = ch_versions.mix(versions)
+
+ emit:
+ ch_resolved
+ ch_versions
+}
+
+def transcriptPatchesArgs(Map config, String method) {
+ def prior_args = ArgsCLI(config.segmentation[method], null, ["prior_shapes_key", "unassigned_value"])
+
+ return ArgsCLI(config.patchify, "micron") + ("comseg" in config.segmentation ? " --write-cells-centroids " : " ") + prior_args
+}
+
+def resolveArgs(Map config) {
+ def gene_column
+ def min_area
+
+ if ("comseg" in config.segmentation) {
+ gene_column = config.segmentation.comseg.config.gene_column
+ min_area = config.segmentation.comseg.min_area ?: 0
+ }
+ else if ("baysor" in config.segmentation) {
+ gene_column = config.segmentation.baysor.config.data.gene
+ min_area = config.segmentation.baysor.min_area ?: 0
+ }
+ else {
+ throw new IllegalArgumentException("Unknown segmentation method in config for resolveArgs")
+ }
+
+ return "--gene-column ${gene_column} --min-area ${min_area}"
+}