generated from statOmics/Rmd-website
-
Notifications
You must be signed in to change notification settings - Fork 5
Expand file tree
/
Copy pathelegansAlignmentCountTable.Rmd
More file actions
191 lines (135 loc) · 5.94 KB
/
elegansAlignmentCountTable.Rmd
File metadata and controls
191 lines (135 loc) · 5.94 KB
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
---
title: "Elegans: Read Alignment and Count Table with QausR and Rhisat2"
author: "Lieven Clement"
date: "statOmics, Ghent University (https://statomics.github.io)"
output:
html_document:
code_download: true
theme: flatly
toc: true
toc_float: true
highlight: tango
number_sections: true
linkcolor: blue
urlcolor: blue
citecolor: blue
---
```{r, echo=FALSE}
suppressPackageStartupMessages({
library(tidyverse)
library(R.utils)
})
```
# Background
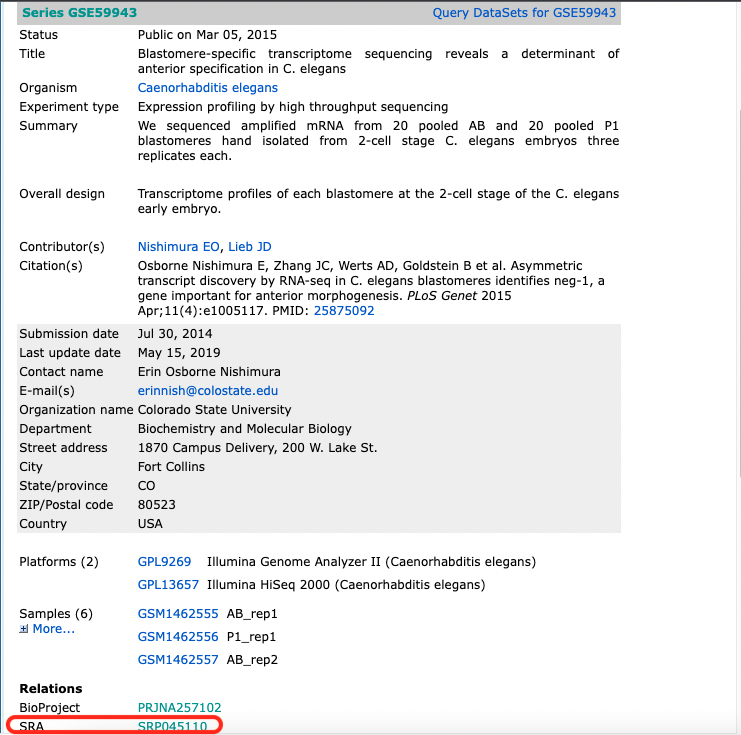
After fertilization but prior to the onset of zygotic transcription, the C. elegans zygote cleaves asymmetrically to create the anterior AB and posterior P1 blastomeres, each of which goes on to generate distinct cell lineages. To understand how patterns of RNA inheritance and abundance arise after this first asymmetric cell division, we pooled hand-dissected AB and P1 blastomeres and performed RNA-seq (Study [GSE59943](https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59943)).
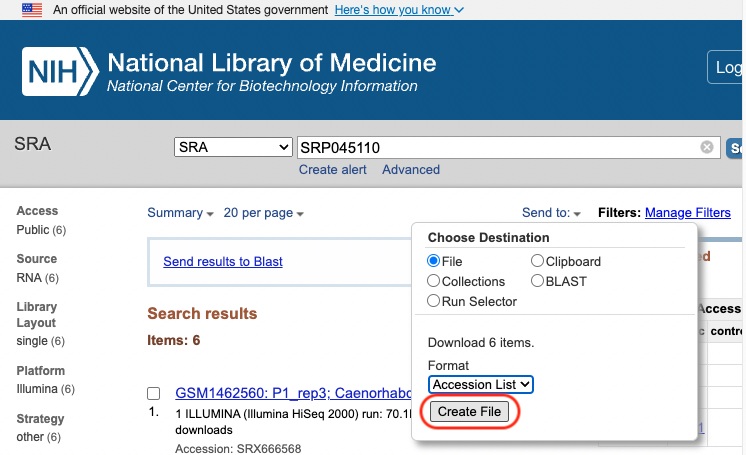
The information which connects the sample information from GEO with the SRA run id is downloaded from [SRA](https://www.ncbi.nlm.nih.gov/sra?term=SRP045110) using the Send to: File button.
# Preprocessing
## Download Data
We used the info in the downloaded SraAccList.txt file to download the SRA files.
We used the system function to invoke aan OS command. xargs can converts each line of a file into an argument.
The fasterq-dump function will then download and convert each corresponding sra file from the SRA archive to a fastq file.
The fasterq-dump is a tool from the [sra-tools](https://github.com/ncbi/sra-tools) suite that can be used to download sra files and to convert them into the fastq format.
```
system("xargs fasterq-dump -p < SraAccList.txt")
```
For illustration purposes we will download subsampled fastq files from the course github site.
```{r}
timeout <- options()$timeout
options(timeout = 300) #prevent timeout
url <- "https://github.com/statOmics/SGA/archive/refs/heads/elegansFastq.zip"
destFile <- "elegansFastq.zip"
download.file(url, destFile)
unzip(destFile, exdir = "./", overwrite = TRUE)
if (file.exists(destFile)) {
#Delete file if it exists
file.remove(destFile)
}
options(timeout = timeout) #prevent timeout
```
## Assess Read Quality
Assess the fastq files with [fastQC](https://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
## Align data
### Input file for hisat aligner
We will make the input file for the hisat aligner that will be called through the QUASAR package.
```{r}
fastqFiles <- list.files(path = "SGA-elegansFastq",full.names = TRUE, pattern = "fastq")
samples <- fastqFiles |>
substr(start=18,stop=27)
fileInfo <- tibble(FileName=fastqFiles,SampleName=samples)
fileInfo
```
We write the fileInfo to disk
```{r}
write_tsv(fileInfo,file="fastQfiles.txt")
```
### Download C. elegans genome and gff3 file
```{r refGenome, echo=FALSE, fig.cap="slide courtesy Charlotte Soneson", out.width='100%'}
knitr::include_graphics("https://raw.githubusercontent.com/statOmics/SGA/master/images_sequencing/humanReferenceGenome_fig.png")
```
```{r refTranscriptome, echo=FALSE, fig.cap="slide courtesy Charlotte Soneson", out.width='100%'}
knitr::include_graphics("https://raw.githubusercontent.com/statOmics/SGA/master/images_sequencing/humanReferenceTranscriptome_fig.png")
```
```{r}
urlGenome <- "https://ftp.ensembl.org/pub/release-108/fasta/caenorhabditis_elegans/dna/Caenorhabditis_elegans.WBcel235.dna.toplevel.fa.gz"
genomeDestFile <- "Caenorhabditis_elegans.WBcel235.dna.toplevel.fa.gz"
urlGff <- "https://ftp.ensembl.org/pub/release-108/gff3/caenorhabditis_elegans/Caenorhabditis_elegans.WBcel235.108.gff3.gz"
gffDestFile <- "Caenorhabditis_elegans.WBcel235.108.gff3.gz"
download.file(url = urlGenome ,
destfile = genomeDestFile)
download.file(url = urlGff,
destfile = gffDestFile)
gunzip(genomeDestFile, overwrite = TRUE, remove=TRUE)
gunzip(gffDestFile, overwrite = TRUE, remove=TRUE)
list.files()
```
### Align reads using QuasR and RHisat package
- We are aligning RNA-seq reads and have to use a gap aware alignment modus. We therefore use the argument `splicedAlignment = TRUE`
- The project is a single-end sequencing project! So we use the argument `paired = "no"`.
```{r}
suppressPackageStartupMessages({
library(QuasR)
library(Rhisat2)
})
sampleFile <- "fastQfiles.txt"
genomeFile <- "Caenorhabditis_elegans.WBcel235.dna.toplevel.fa"
projElegans <- qAlign(
sampleFile = sampleFile,
genome = genomeFile,
splicedAlignment = TRUE,
aligner = "Rhisat2",
paired="no")
```
## Make count table
### Convert gff file into database
The gff3 file contains information on the position of features, e.g. exons and genes, along the chromosome.
```{r}
suppressPackageStartupMessages({
library(GenomicFeatures)
library(Rsamtools)
})
annotFile <- "Caenorhabditis_elegans.WBcel235.108.gff3"
#chrLen <- scanFaIndex(genomeFile)
#chrominfo <- data.frame(chrom = as.character(seqnames(chrLen)),
# length = width(chrLen),
# is_circular = rep(FALSE, length(chrLen)))
txdb <- makeTxDbFromGFF(file = annotFile,
dataSource = "Ensembl",
organism = "Caenorhabditis elegans")
```
### Make Gene Count table
With the qCount function we can count the overlap between the aligned reads and the genomic features of interest.
```{r}
geneCounts <- qCount(projElegans, txdb, reportLevel = "gene")
head(geneCounts)
```
Save count table for future use.
```{r}
write.csv(as.data.frame(geneCounts),file = "elegansCountTable.csv")
```
# Session Info
With respect to reproducibility, it is highly recommended to include a session info in your script so that readers of your output can see your particular setup of R.
```{r}
sessionInfo()
```