![]()




This software is currently in ALPHA. While we have extensively tested these tools across a wide variety of vendor-provided data, no guarantees are made regarding correctness or stability.
We are targeting June 1, 2026 to recommend fgumi over fgbio for production use.
Fulcrum Genomics Unique Molecular Indexing (UMI) Tools - a suite of high-performance tools for working with UMI-tagged sequencing data.

fgumi provides comprehensive functionality for:
- UMI extraction from FASTQ files
- Read grouping by UMI with multiple assignment strategies
- UMI-aware deduplication for marking/removing PCR duplicates
- Consensus calling (simplex, duplex, and CODEC)
- Quality filtering of consensus reads
- Read clipping for overlapping pairs
- Metrics collection for QC and analysis
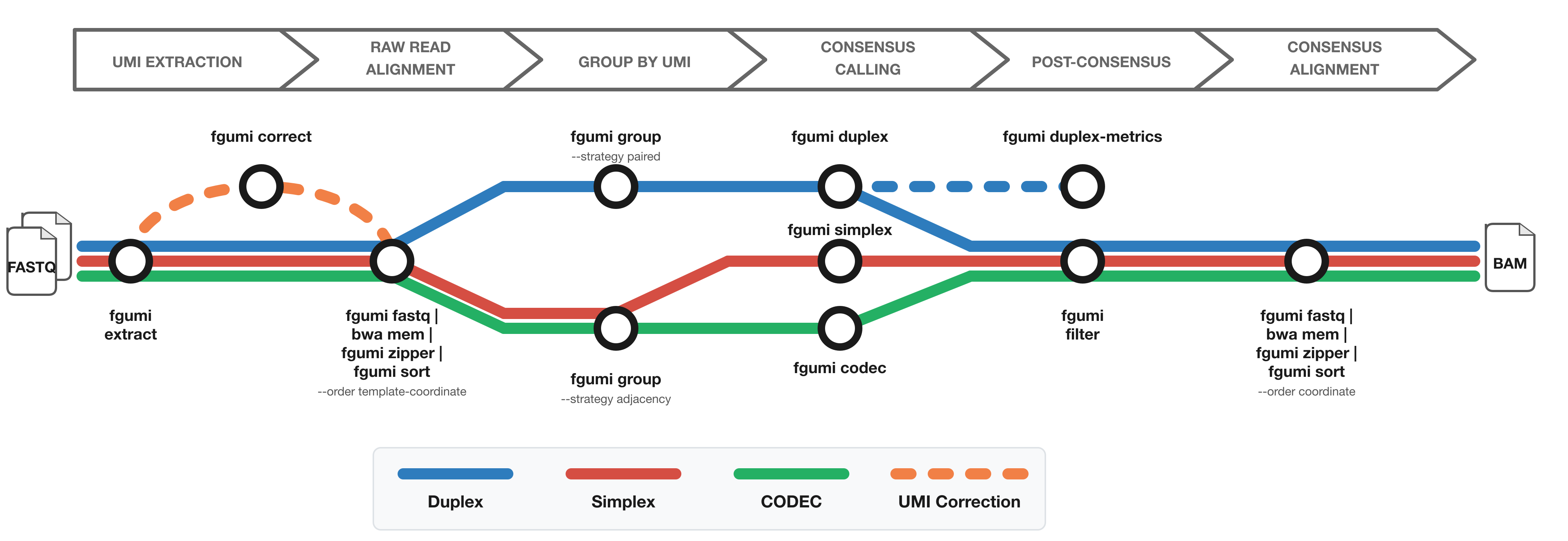
The diagram shows the workflow from FASTQ files to filtered consensus reads:
- Red: Simplex (single-strand) consensus
- Blue: Duplex (double-strand) consensus
- Green: CODEC consensus
- Orange: Optional UMI correction for fixed UMI sets
- Documentation
- Best Practice Pipeline: Recommended workflow from FASTQ to consensus
- Performance Tuning Guide: Threading, memory, and compression optimization
- Snakemake Pipeline: Reference implementation
- Metrics: Output metrics documentation
- Developing: Developer guide
- Compare CLI: Compare command documentation (feature-gated)
- Simulate CLI: Simulate command documentation (feature-gated)
- Releases
- Issues: Report a bug or request a feature
- Pull requests: Submit a patch or new feature
- Discussions: Ask a question
- Contributors guide
- License: Released under the MIT license
Pre-built binaries for the most common operating systems and CPU architectures are attached to each release for this project.
cargo install fgumi
Clone the repository:
git clone https://github.com/fulcrumgenomics/fgumi
Build the release version:
cd fgumi
cargo build --release
| Feature | Description |
|---|---|
compare |
Developer tools for comparing BAMs and metrics |
simulate |
Commands for generating synthetic test data |
profile-adjacency |
Enable profiling output for adjacency UMI assigner |
Enable with: cargo build --release --features <feature> |
| Command | Description | Equivalent Tool(s) |
|---|---|---|
extract |
Extract UMIs from FASTQ files | fgbio ExtractUmisFromBam |
correct |
Correct UMIs based on sequence similarity | fgbio CorrectUmis |
zipper |
Restore original FASTQ from unaligned BAM | fgbio ZipperBams, picard MergeBamAlignment |
fastq |
Convert BAM to FASTQ format | samtools fastq |
sort |
Sort BAM by coordinate/queryname/template | — |
group |
Group reads by UMI | fgbio GroupReadsByUmi |
dedup |
Mark/remove UMI-aware duplicates | gatk UmiAwareMarkDuplicatesWithMateCigar, umi-tools dedup |
simplex |
Call single-strand consensus reads | fgbio CallMolecularConsensusReads |
duplex |
Call duplex consensus reads | fgbio CallDuplexConsensusReads |
codec |
Call CODEC consensus | fgbio CallCodecConsensusReads |
filter |
Filter consensus reads | fgbio FilterConsensusReads |
clip |
Clip overlapping read pairs | fgbio ClipBam |
duplex-metrics |
Collect duplex metrics | fgbio CollectDuplexSeqMetrics |
review |
Review consensus variants | fgbio ReviewConsensusVariants |
downsample |
Downsample BAM by UMI family | N/A |
compare <cmd> |
Compare files (feature-gated) | N/A |
simulate <cmd> |
Generate test data (feature-gated) | N/A |
For detailed usage of each command, run:
fgumi <command> --help- Extract UMIs from FASTQ:
fgumi extract \
--inputs R1.fastq.gz R2.fastq.gz \
--read-structures +T +M \
--output unaligned.bam \
--sample MySample \
--library MyLibrary- (Optional) Correct UMIs for fixed UMI sets:
fgumi correct \
--input unaligned.bam \
--output corrected.bam \
--umi-files umis.txt \
--min-distance 1- Align and sort reads using fgumi fastq + zipper + sort pipeline:
fgumi fastq --input unaligned.bam \
| bwa mem -p ref.fa - \
| fgumi zipper --unmapped unaligned.bam \
| fgumi sort --output sorted.bam --order template-coordinate- Group reads by UMI:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy paired # for duplex workflows
# or --strategy adjacency for simplex/codec workflows- Call consensus reads:
# Simplex consensus
fgumi simplex \
--input grouped.bam \
--output consensus.bam
# Or duplex consensus
fgumi duplex \
--input grouped.bam \
--output duplex.bam
# Or codec consensus
fgumi codec \
--input grouped.bam \
--output codec_consensus.bam- (Optional) Collect duplex metrics:
fgumi duplex-metrics \
--input grouped.bam \
--output metrics- Filter consensus reads:
fgumi filter \
--input consensus.bam \
--output filtered.bam \
--ref ref.fa \
--min-reads 1,1,1fgumi supports multi-threading and memory management for optimal performance:
- Threading:
--threads Nfor parallel processing - Memory:
--queue-memory 768(plain numbers are MB; supports human-readable formats like2GB) - Compression:
--compression-level 1-12for speed vs size trade-offs
Memory Model — Important for fgbio users: Unlike fgbio's JVM
-Xmxwhich sets a hard ceiling on total process memory, fgumi's--queue-memorycontrols pipeline queue backpressure only. Two things to be aware of:
- Per-thread scaling (default):
--queue-memory 768 --threads 8allocates 768 MB per thread = ~6 GB total queue memory. Use--queue-memory-per-thread falsefor a fixed total budget.- Queue memory < total process memory: Actual RSS will be higher due to UMI data structures, decompressors, thread stacks, and working buffers.
See the Performance Tuning Guide for detailed guidance, including scenario-based configurations and troubleshooting.
fgumi is written in Rust for maximum performance.
| Command | Key Optimizations |
|---|---|
extract |
Work-stealing thread pool, streaming I/O |
correct |
N-gram indexing with pigeonhole principle, BK-tree for k>1 |
group |
2-bit UMI encoding, N-gram/BK-tree indexing, directed adjacency graph |
simplex |
Fast-path for unanimous consensus, parallel processing |
duplex |
Parallel duplex calling, efficient strand matching |
codec |
Parallel CODEC consensus |
filter |
Streaming filter with parallel processing |
clip |
Parallel overlap detection and clipping |
sort |
External merge sort, configurable memory limit |
- 2-bit DNA encoding: 4 bases in 1 byte, 32 bases in u64
- CPU intrinsics: XOR + popcount for Hamming distance
- Work-stealing scheduler: Unified pipeline with dynamic load balancing
- libdeflate: Fast BGZF compression
fgumi's UMI grouping algorithms are inspired by:
- UMI-tools (Smith et al. 2017) - The directed adjacency method for UMI deduplication with count gradient constraints.
- UMICollapse (Liu 2019) - N-gram and BK-tree indexing strategies for efficient similarity search in UMI deduplication.
Development of fgumi is supported by Fulcrum Genomics.
This software is under active development. While we make a best effort to test this software and to fix issues as they are reported, this software is provided as-is without any warranty (see the license for details). Please submit an issue, and better yet a pull request as well, if you discover a bug or identify a missing feature. Please contact Fulcrum Genomics if you are considering using this software or are interested in sponsoring its development.