Manual
NGSphy documentation: v 20171214
© 2017 Merly Escalona (merlyescalona@uvigo.es), Sara Rocha, David Posada
University of Vigo, Spain, http://darwin.uvigo.es
NGSphy is a Python open-source tool for the genome-wide simulation of NGS data (read counts or Illumina reads) obtained from thousands of gene families evolving under a common species tree, with multiple haploid and/or diploid individuals per species, where sequencing coverage (depth) heterogeneity can vary among species, individuals and loci, including off target or uncaptured loci.
If you use NGSphy, please cite:
- Escalona, M, Rocha S and Posada D. (2018) NGSphy: phylogenomic simulation of NGS data. Bioinformatics; doi: https://doi.org/10.1093/bioinformatics/bty146
- Sukumaran, J and Holder MT. (2010). DendroPy: A Python library for phylogenetic computing. Bioinformatics 26: 1569-1571.
if running ART cite also:
- Huang W, Li L, Myers JR and Marth, GT. (2012) ART: a next-generation sequencing read simulator. Bioinformatics 28 (4): 593-594
if using SimPhy cite also:
- Mallo D, De Oliveira Martins L and Posada D. (2016). SimPhy : Phylogenomic Simulation of Gene, Locus, and Species Trees. Systematic Biology 65(2): 334-344.
If using single gene tree inputs, cite also:
- Fletcher, W and Yang Z. (2009) INDELible: A flexible simulator of biological sequence evolution. Molecular Biology and Evolution 26 (8): 1879–88.
[Single gene-tree scenario]
- NGSPhy settings file
- INDELible control file
- Newick file with single gene tree
- ancestral sequence file (FASTA) (optional)
- reference allele file (optional)
[Species-tree scenario]
- NGSPhy settings file
- SimPhy output
- reference allele file (optional)
- coverage variation
- CSV
- plain text
- log files
- bash scripts
NGSphy allows to introduce coverage heterogeneity for three different kind of scenarios, following statistical distributions (more details on Manual - Section 7.3):
-
Experiment coverage: allows to introduce expected coverage for each replicate, and the variation of coverage across individuals and across loci.
- Available for all input/simulation modes.
-
Targeted sequencing parameters: allow the user to emulate the variation in depth of coverage that can occur in a targeted-sequencing experiment. These parameters identify the on target/off target loci as well as the number of loci that may not be captured.
- Only possible for input mode 4 - gene tree distributions (SimPhy data).
-
Taxon-specific effects: allows the user to define of coverage variation for specific taxa. It can be used for example to emulate a decay in coverage, related to the phylogenetic distance of the a species to the reference species used to build the target-loci probes.
- Available for all input/simulation modes.
NGSphy has been developed for Linux/MAC environments with Python 2.7.
To install NGSphy you need to:
- clone its git repository and download the required third-party software (Section 5.3):
# 1. Clone NGSphy repository
git clone https://github.com/merlyescalona/ngsphy.git
# 2. Move to ngsphy folder
cd ngsphy
# 3. Extract files and install
sudo python setup.py install # if user has sudoer permissions
# python setup.py install --user # if user does not have permissions as sudoer
- or install NGSphy through
pipand download the required third-party software (Section 5.3):
sudo pip install ngsphy # if user has sudoer permissions
# pip install --user ngpshy # if user does not have permissions as sudoer
This should allow the user to call ngsphy executable (in the scripts folder). In case this does not happen modify the execution permissions:
# 1. go to the scripts folder
cd ngsphy/scripts
# 2. modify the execution permissions
chmod +x ngsphy
ART is a set of simulation tools to generate synthetic next-generation sequencing reads. You can download it from:
http://www.niehs.nih.gov/research/resources/software/biostatistics/art/
Version ChocolateCherryCake or later.
Following installation instructions from ART, you can download the binaries or compile the source code. If you decide to compile the source code:
# 1. Extract files from the compressed tgz
cd /path/to/art-download
tar -xvf artsrcmountrainier20160605linuxtgz.tgz
# 2. Change current directory to the extracted one
cd art_src_MountRainier_Linux/
# 3. Make sure you have all the dependencies installed and generate the Makefile
./configure
# 4. Run the Makefile
make
INDELible is an application for sequence simulation. You can download it from:
Version 1.03.
In order to get INDELible, you will need to register. It is free software, and is distributed under: GNU General Public License as published by the Free Software Foundation, either version 3 of the License, or any later version. For more information go to http://www.gnu.org/licenses/.
Once the software is downloaded:
- Unpack the archive on Unix-like systems using:
# 1. Change directory to the download folder.
cd /path/to/indelible-download
# 2. Extract file from the compressed file.
tar -xvzf INDELibleV1.03.tar.gz
-
(a) If you want to compile from source:
- Move to the source folder:
# 3a. Move to the source folder cd INDELibleV1.03/src/- Include the following line at to the top of MersenneTwister.h file.
#include <unistd.h>- Compile INDELible using:
# 4a.Compile g++ -o indelible indelible.cpp -lm -
(b) If you want to use the binaries directly :
# This is an example for MacOS
# 3b. Move to the binaries folder
cd INDELibleV1.03/bin/
# 4b. Rename the binary (for proper NGSphy execution)
mv indelible_1.03_OSX_intel indelible
# 5b. Modified execution permissions
chmod +x indelible
This is version of INDELible we have modified to allow the use of a given ancestral sequence as root. It can be obtained from cloning its repository:
- (a) If you want to compile it:
# 1. Clone the repository
git clone https://github.com/merlyescalona/indelible-ngsphy.git indelible-ngsphy
# 2. Change directory to indelible-ngsphy source code folder.
cd indelible-ngsphy/
# 3. Compile
make
- (b) If you want to use the binaries directly :
# This is an example for MacOS
# 3b. Move to the binaries folder
cd indelible-ngsphy/bin/
# 4b. Rename the binary (for proper NGSphy execution)
mv indelible-ngsphy_OSX_intel_i386 indelible-ngsphy
# 5b. Modified execution permissions
chmod +x indelible-ngsphy
SimPhy can be obtained from cloning its repository and installing its dependencies. Detailed, information on how to install SimPhy here.
# 1. Clone the repository
git clone https://github.com/adamallo/SimPhy.git
Once all software has been installed, it must be added to the path.
First you have to add the lines below to the ~/.bashrc (or ~/.bash_profile according to user's environment) file to keep the changes permanently.
ART="/path/to/art/executable"
INDELIBLE="/path/to/indelible/executable"
INDELIBLENP="/path/to/indelible-ngsphy/executable"
NGSPHY="/path/to/ngsphy/scripts/"
SIMPHY="/path/to/simphy/executable"
export PATH="$ART:$INDELIBLE:$INDELIBLENP:$NGSPHY:$SIMPHY:$PATH"
Apply changes:
source ~/.bashrc
# source ~/.bash_profile
NGSphy does not have a Graphical User Interface (GUI) and works on the Linux/Mac command line in a non-interactive fashion.
usage: ngsphy [-s <settings_file_path>]
[-l <log_level>] [-v] [-h]
-
Optional arguments:
-
-s <settings_file_path>, --settings <settings_file_path>:- Path to the settings file. This is optional, by default NGSphy loos for a
settings.txtfile in the current working directory. You can also specify a particular settings file with:
ngsphy -s my_settings.txt - Path to the settings file. This is optional, by default NGSphy loos for a
-
-l <log_level>, --log <log_level>: Specified hierarchical log levels that will be shown through the standard output. A detailed log will be stored in a separate file. Possible values:-
DEBUG: shows very detailed information of the program's process. -
INFO(default): shows only information about the state of the program. -
WARNING: shows only system warnings. -
ERROR: shows only execution errors.
-
-
-
Information arguments:
-
-v, --version: Show program's version number and exit. -
-h, --help: Show help message and exit.
-
NOTE: Examples of the settings file can be found under the
data/settingsfolder in the NGSphy source.
NGSphy requires a settings file “settings.txt” that specifies the different options and parameter values for the simulations. A settings file with a different name can be specified with the -s/--settings option. The information in the settings file is organized in 6 optional/required blocks:
-
[general]: general parameters. -
[data]: specifies the type of input data as well as input parameters and files. -
[coverage]: parameters that describe the variation of coverage in the dataset (optional). -
[ngs-reads-art]: specifies ART execution parameters (optional) -
[ngs-read-counts]: specifies parameters for read counts (optional). -
[execution]: describes how the execution of the whole process will be made (optional).
Stores general parameters for each NGSphy run.
[general]
path=/home/user/
output_folder_name=NGSphy_output
ploidy=1
-
path:- purpose: path where output folder will be created.
- type: string (path).
-
output_folder_name:- purpose: name of the output folder where NGSphy results will be stored. If the output folder already exists, the new output folder will get the same base name with a numerical suffix (
outputFolder_n), representing the nth time the program with that output folder name was ran. - type: string.
- value:
NGSphy_output(default).
- purpose: name of the output folder where NGSphy results will be stored. If the output folder already exists, the new output folder will get the same base name with a numerical suffix (
-
ploidy:- purpose: refers to the ploidy that the resulting individuals will have. So far it is only possible to generate haploid and diploid individuals.
- type: number (integer).
- values: value in the closed-interval
[1,2](default1).
-
seed:- purpose: random number generator seed.
- type: number (integer).
- values: value in the closed-interval [0, 2^32 - 1]
Defines the input data for NGSphy, which consists of 4 different modes:

FIGURE 1: Input modes: a) a single gene tree; b) single gene tree with a user-defined ancestral sequence; c) a single gene tree with an anchor sequence and d) gene-tree distributions (SimPhy output [species-tree simulations])
Single gene tree
[data]
inputmode=1
gene_tree_file=/home/myuser/my_gene_tree.tree
indelible_control_file=/home/myuser/my_control_indelible.txt
Single gene tree with user-defined ancestral sequence
[data]
inputmode=2
gene_tree_file=/home/myuser/my_gene_tree.tree
ancestral_sequence_file=/home/myuser/my_ancestral.fasta
indelible_control_file=/home/myuser/my_control_indelible.txt
Single gene tree with user-defined anchor sequence
[data]
inputmode=3
gene_tree_file=/home/myuser/my_gene_tree.tree
anchor_sequence_file=/home/myuser/my_anchor.fasta
anchor_tip_label=1_0_0
indelible_control_file=/home/myuser/my_control_indelible.txt
Gene-tree distribution - SimPhy output (species-tree simulations)
[data]
inputmode=4
simphy_folder_path=testSimphy
simphy_data_prefix=data
simphy_filter=true
-
inputmode:- purpose: identifies the type of input.
- type: number (integer)
- value: values within the closed interval
[1,4]- single gene tree.
- single gene tree with an ancestral sequence.
- single gene tree with an anchor sequence.
- gene-tree distribution (SimPhy output [species-tree simulations]).
-
gene_tree_file- purpose: path of the gene tree in Newick format. There must be a single path and a single tree in the file.
The name of the file, without extension, must be the same as the name of the tree within the INDELible control file, in the
[NGSPHYPARTITION]option. - type: string (path).
- format: see specification in Section 7.2.6. (INDELible control file).
- purpose: path of the gene tree in Newick format. There must be a single path and a single tree in the file.
The name of the file, without extension, must be the same as the name of the tree within the INDELible control file, in the
-
indelible_control_file- purpose: path for the INDELible control file.
- type: string (path).
- format: see specification in Section 7.2.6. (INDELible control file).
These options are related to the INDELible run with a user-defined ancestral sequence:
-
gene_tree_file- purpose: same as in Section 7.2.2. (Single gene tree).
- type: string (path).
-
ancestral_sequence_file- purpose: path to the FASTA file that contains the ancestral sequence.
- type: string (path).
-
indelible_control_file- purpose: Same as Section 7.2.2. (Single gene tree).
- type: string (path).
These options are related to the INDELible run with a user-defined ancestral sequence:
-
gene_tree_file- purpose: same as in Section 7.2.2. (Single gene tree).
- type: string (path).
-
anchor_sequence_file- purpose: path to the FASTA file that contains the anchor sequence.
- type: string (path).
-
anchor_tip_label- purpose: tip label of the gene tree that corresponds to the tip that will be used as root.
- type: string.
- format: see specification in the Section 7.2.7. (Single gene-tree file labeling).
-
indelible_control_file- purpose: Same as Section 7.2.2. (Single gene tree).
- type: string (path).
If the user wants to use a tip sequence as the root for the alignment simulation (anchor_sequence),
the gene tree has to be re-rooted (anchor_tip_label), so that simulation can proceed using the anchor_sequence as the root node.
In the example shown in Figure 1, NGSphy would transform the tree on the left into the one on the right, using as anchor tip 2_0_0.
The key observation here is that the branch length from node A to tip 2_0_0 has to become zero. Then, the re-rooted tree plus the anchor (known)
sequence are given to indelible-ngsphy, with the INDELible control file (format in
Section 7.2.6. (INDELible control file)) to simulate the corresponding sequence alignments under the model from the control file.
 FIGURE 2: Re-rooting process. Number above branches indicate branch length.
FIGURE 2: Re-rooting process. Number above branches indicate branch length.
-
simphy_folder_path- purpose: path to the folder with SimPhy’s output.
- type: string (path).
-
simphy_data_prefix- purpose: prefix used in SimPhy's run.
- type: string.
-
simphy_filter [optional]- purpose: filter out the replicates that do not satisfy the required ploidy. For the diploid case the number of gene tree tips per species has to be an even number. See more in Section 7.2.8. (Individual assignment).
- type: boolean.
- value:
-
0,false,off: don’t filter (default). -
1,true,on: filter.
-
A detailed description of SimPhy's output can be found in https://github.com/adamallo/simphy. The SimPhy output required by NGSphy has to include:
-
<simphy_project_name>.command: a plain text file with the original command line arguments. -
<simphy_project_name>.db: a SQLite database composed by3linked tables with different information about species, locus and gene trees. -
<simphy_project_name>.params: a plain text file summarizing the sampled options. - a set of folders with the multiple sequence alignments and the corresponding trees.
When the input mode is a single gene tree, it is necessary to have a control file to call INDELible. Here, we use a slightly modified version of the INDELible's control file. To properly set up the configuration file for INDELible, users should refer first to INDELible’s manual. In our version, the file must include the following blocks:
-
[TYPE]: 1 block -
[SETTINGS]: 1 block (optional) -
[MODEL]: 1 block -
[NGSPHYPARTITION]: 1 block
NOTE: Including a wrong number of blocks or other type of blocks will result in an error message and will terminate NGSPhy execution.
-
[TYPE]standard INDELible specification. -
[SETTINGS]standard INDELible specification. -
[MODEL]standard INDELible specification. -
[NGSPHYPARTITION]this block defines:- the gene tree for INDELible (this name has to be the same as the Newick file used as input (see Section 7.2).
- the substitution model for INDELible. This name must match the name of the model used in the previous
[MODEL]block. - the sequence length.
For example, we have a gene tree in the Newick file: tree1.tree, where sequences will evolve under model m1, with a length of 500bp.
[NGSPHYPARTITION] tree1 m1 500
NOTE: Examples of the modified INDELible control files can be found under the
data/indeliblefolder in the NGSphy source.
Single gene trees in Newick format should have specific tip labels. Tips must follow a specific format in order to be managed by NGSphy. This format indicates species, locus and individual with the scheme (X_Y_Z) where:
-
X stands for the the species identifier, where
X > 0 -
Y for the locus identifier, where
Y > 0 -
Z for the individual identifier, where
Z > 0
The gene tree file must be in Newick format, rooted and with branch lengths. If the gene tree is not rooted, it will be forced following Dendropy specifications.
For example, if we have 3 species and 2 gene copies per species the labels would be:

FIGURE 3: Gene tree labeling example.
(((1_0_1:1.0,1_0_0:2.0):1.0, (2_0_1:1.0,2_0_0:1.0)),( (3_0_1:2.0,3_0_0:3.0) ));
For haploid individuals, each tip in the gene tree provided will correspond to a single individual. For diploid individuals the number of gene-tree tips per species must be even. In this case, the individuals are generated by randomly sampling without replacement two gene copies from a specific gene-family until all gene tree tips have been assigned to an individual.
For the gene-tree distribution input mode only, the outgroup in the gene trees is
called 0_0_0 and has one gene copy. Therefore, for the generation of diploid individuals,
the outgroup will be homozygous, obtained by the duplication of the sequence of its gene copy.
The description of the sequence(s) in the final FASTA file of the individual is formatted as:
>project:repID:locusID:sequence_file_prefix:indID:full_sequence_description
Where:
-
project: if using any of the single gene tree input modes, it will be NGSphy. For the gene-tree distribution input mode, it will be the name of the SimPhy output folder. -
repID: replicate identifier. -
locID: locus identifier. -
indID: individual identifier. -
sequence_file_prefix: if using any of the single gene tree input modes, it will be ngsphydata. For the gene-tree distribution input mode, it will be the simphy_data_prefix. -
full_sequence_description: the description of the original sequence.
Sequencing coverage can be specified at three different levels: experiment, individual and locus-wide. It is also possible to mimic the variation in coverage expected for targeted sequencing, including off target loci and taxon-specific effects.
[coverage]
experiment=F:100
individual=LN:1.2,1
locus=LN:1.3,1
offtarget=0.4, 0.01 # 40% loci are off target, will have 1% of the coverage
notcaptured=0.5
taxon= 1,0.5;2:0.25
The parameters that will define the coverage in NGSphy have to be provided using a specific notation in order to define statistical distributions and dependency between arguments. The sampling notation is structured as a particular statistical distribution (see code for the statistical distribution), followed by a colon and a list of comma-separated parameter values:
distribution_code:param1,param2, ...
For example:
- Fixed value=100.
F:100
- Poisson distribution with mean=100.
P:100

FIGURE 4: Sampling notation example. Poisson distribution.
- Negative Binomial, mean=100 and overdispersion=10.
NB:100,10

FIGURE 5: Sampling notation example. Negative Binomial distribution.
| Distribution | Code | Num. parameters | Parameters | Description |
|---|---|---|---|---|
| Binomial | b/B |
2 |
r,p | trials, probabilities |
| Exponential | e/E |
1 |
s | scale |
| Fixed Point | f/F |
1 |
v | value |
| Gamma | g/G |
2 |
sh,sc | shape, scale |
| Log Normal | ln/LN |
2 |
mu,sd | mean, standard deviation |
| Negative Binomial | nb/NB |
2 |
mu,r | mean of the underlying Poisson distribution, overdispersion |
| Normal | n/N |
2 |
mu,var | mean, variance |
| Poisson | p/P |
1 |
mu | mean |
| Uniform | u/U |
2 |
min,max | minimum (included), maximum (excluded) |
-
experiment- purpose: expected depth of coverage for a specific replicate.
- type: fixed value or statistical distribution.
-
locus [optional]- purpose: variation of expected coverage between loci.
- type: fixed value or statistical distribution.
-
individual [optional]- purpose: variation of expected coverage between individuals.
- type: fixed value or statistical distribution.
-
offtarget [optional]- purpose: related to targeted-sequencing experiments; percentage of loci that will be considered off target (captured and sequenced but not originally targeted); expected coverage will be a proportion of the sampled coverage.
- type: 1 pair (
proportionLoci,proportionCoverage). - values:
-
proportionLoci: number (float) in the closed interval
[0,1]. -
proportionCoverage: number (float) in the closed interval
[0,1].
-
proportionLoci: number (float) in the closed interval
-
notcaptured [optional]- purpose: related to targeted-sequencing experiments; fraction of originally targeted loci that will not be captured/sequenced.
- type: number (float).
- value: number in the closed interval
[0,1].
-
taxon [optional]- purpose: related to targeted-sequencing experiments; decrease in coverage for particular species. It can be due to the phylogenetic distance between a reference species (used to design the probes for the targeted loci) and the individuals from the target-sequencing experiment or to species-specific sample conditions.
- type: pairs (
speciesID,coverageProportion) - values:
- speciesID: one or more of the existent species in the tree.
-
coverageProportion: value in the closed interval
[0,1].
- format:
taxon=speciesID1,coverageProportion1; speciesID2,coverageProportion2 ...
The experiment-wide coverage is sampled for each replicate from the specified statistical distribution,
and this value becomes the expected coverage for every loci and individual in that replicate. For example,
if experiment=P:100, we might sample a value of 104 for replicate 1, so the expected coverage would
be 104x for that particular experiment.
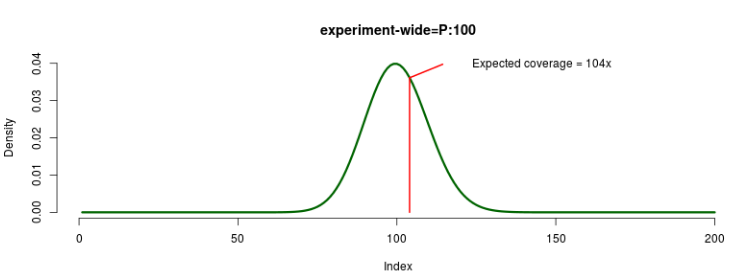
FIGURE 6: Experiment-wide coverage sampling example.
An individual-wide coverage multiplier is sampled for each individual within a given replicate.
The value indicated in the settings file is in fact a hyper-parameter that controls a specific hyper-distribution from
which a single value is sampled per replicate. For that replicate, this value will become the shape of a Gamma
distribution with mean = 1, from which a multiplier is sampled for each individual.
In exactly the same manner, a locus-wide coverage multiplier is sampled for each locus within
a given replicate. The value indicated in the settings file is again a hyper-parameter that controls
a specific hyper-distribution from which a single value is sampled per replicate. For that replicate,
this value will become the shape of a Gamma distribution with mean = 1, from which a multiplier is sampled for each loci.
For example, imagine we have 2 replicates, 2 loci, 2 individuals and input the following coverage settings:
[coverage]
experiment: P:100
locus: LN:1.2,1
individual: E:1
First, we sample from a Poisson, with mean=100, to obtain the expected coverage per experiment (rep1c, rep2c).

FIGURE 7: Experiment-wide coverage sampling a complex example.
Coverage variation before locus/individual multipliers:
| Replicate | Locus | Expected Coverage | |
| Individual I | Individual II | ||
| 1 | A | 102 | 102 |
| B | 102 | 102 | |
| 2 | A | 112 | 112 |
| B | 112 | 112 | |
Afterwards, we sample the locus-wide rate multipliers from the hyper-distribution, in
this case a Log Normal with mean = 1.2 and standard deviation = 1 (locrep1 a,locrep2 a).
This, give us the shape of the Gamma distribution with mean = 1 from which we sample the
rate multipliers, as many as loci (locAm, locBm).
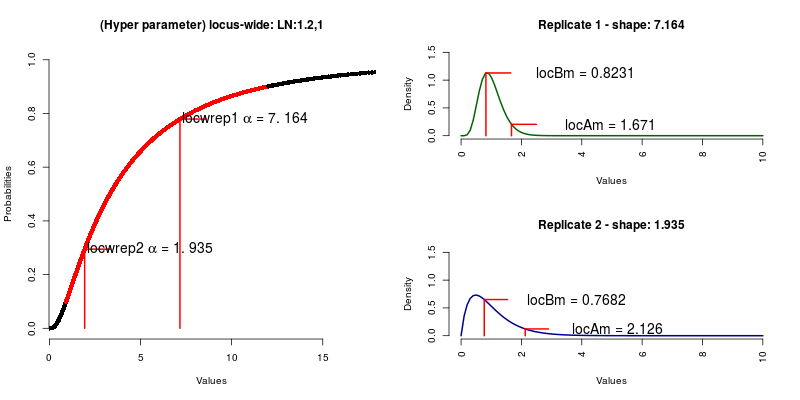
FIGURE 8: Locus-wide coverage sampling.
Coverage variation after locus-wide multipliers:
| Replicate | Locus | Rate Multiplier (per loci) | Resulting coverage Coverage | |
| Individual I | Individual II | |||
| 1 | A | 1.671 | 170.4420 | 170.4420 |
| B | 0.8231 | 83.9562 | 83.9562 | |
| 2 | A | 2.1260 | 238.1120 | 238.1120 |
| B | 0.7682 | 86.0384 | 86.0384 | |
Next, we get the individual-wide rate multipliers, sampling from the hyper-distribution,
an Exponential with rate = 1 (indwrep1 a, indwrep2 a). This, give us the shape of the
Gamma distribution with mean = 1 from which we sample the rate multipliers, as many as individuals (indAm, indBm).

FIGURE 9: Individual-wide coverage sampling.
| Replicate | Individuals | Rate Multiplier (per individual) | Resulting coverage Coverage | |
| Locus A | Locus B | |||
| 1 | I | 0.4849 | 82.64733 | 40.71036 |
| II | 1.679 | 286.1721 | 140.9625 | |
| 2 | I | 0.7437 | 177.08389 | 63.98676 |
| II | 1.325 | 315.4984 | 114.0009 | |
Targeted sequencing parameters allow the user to emulate the variation in depth of
coverage that can occur in a targeted-sequencing experiment. This is possible when using
gene tree distributions (SimPhy project) as input data. These parameters identify the
on target/off target loci as well as the number of loci that may not be captured. While
on target loci will keep their expected coverage, the off target fraction will have a
(user-defined) fraction of this. The not-captured indicates the fraction of targeted
loci that will not be captured, and its expected coverage will be 0x. For example,
if we have 2 replicates, 3 loci, and input the following coverage:
[coverage]
experiment-wide: P:100
off target=0.33 0.1
notcaptured=0.5 # half of the on target
If we consider the same coverage sampling as before, P:100:
| Replicate | Locus | Category | Expected coverage | Rate Multiplier | Sampled coverage |
| 1 | A | on target | 105 | 1 | 105 |
| B | on target, not captured | 105 | 0 | 0 | |
| C | off target | 105 | 0.1 | 10.5 | |
| 2 | A | on target | 92 | 1 | 92 |
| B | on target, not captured | 92 | 0 | 0 | |
| C | off target | 92 | 0.1 | 9.2 |
Taxon-specific effects allows the user to define of coverage variation for specific taxa. It can be used for example to emulate a decay in coverage, related to the phylogenetic distance of the a species to the reference species used to build the target-loci probes (Bragg et al, 2016) (this is sometimes called phylogenetic decay) or in a more general context for particular sample conditions (low amount of DNA, museum specimens, etc. ).

FIGURE 10: Taxon-specific coverage explanation.
For example:
[coverage]
experiment: F:60
taxon=1,0.5; 2,0.25
Meaning that, if the expected coverage for the experiment is 60x, individuals from the species speciesID = 1,
will have a coverage of 30x (50% of the expected coverage) and the individuals from the species speciesID = 2,
will have coverage of 15x (25% of the expected coverage).
Defines the options for ART.
If the user specifies here any input (in,i) or output (out,o) arguments, these will be ignored since
these values will be auto-generated.
For coverage related options, keep in mind that there are 2 different situations:
-
-c,--rcount: number of reads/read pairs to be generated per sequence/amplicon (not be used together with -f/--fcov) -
-f,--fcov: the fold of read coverage to be simulated or number of reads/read pairs generated for each amplicon.
Here, these parameters will be treated as boolean (true,false) while its expected value will be set with the
istributions given in the [coverage] block. Units will be
reads or depth of coverage according to the ART option selected.
| Parameter | Value | Units |
|---|---|---|
| -c, --rcount | True/on/1 | reads |
| -f, --fcov | False/off/0 | depth (x) |
IMPORTANT: In NGSphy the coverage is defined per individual not per sequence/amplicon.
NGSphy creates two sets of VCF files (a VCF file per locus per replicate) that contain contains the variable positions and their genotype likelihoods calculated upon reads with/without sequencing errors. The latter VCF therefore also unveils the true genotypes (those with non-zero genotype likelihoods).
[ngs-read-counts]
read_counts_error=0.1
reference_alleles_file=/home/myuser/my_reference_alleles.txt
-
read_counts_error- purpose: to emulate sequencing error.
- type: number (float).
- value: value in the left-closed interval
[0,1).
-
reference_alleles_file- purpose: identifiers of the sequences used as reference for the variable sites.
- type: string (path).
Defines which alleles will be used as references to generate the VCF files. The description of the allele sequences follow the labeling explained above in Section 7.2.7 (Single gene-tree file labeling). The content of the file should be formatted as:
repID, spID,locID, indID
Where:
-
repID, replicate ID. -
spID, species ID (Xvalue of the sequence description) -
locID, locus ID (Yvalue of the sequence description) -
indID, gene tree tip ID (Zvalue of the sequence description).
IMPORTANT: By default, if the reference allele file is not specified (or badly formatted), the reference allele will correspond to the sequence named
1_0_0.
The simplest case will be when the input is a single tree and all individuals have the same
number of loci. So, let’s suppose we want to run NGSphy, with single gene tree inputmode (inputmode = 1).
The gene tree is the following (as in Section 7.2.7. (Single gene-tree file format and labeling)):
FIGURE 11: Example of gene tree with proper label notation.
Also, we want to generate read counts, with no errors, and we want to use the gene-tree tip with label 2_0_1 as the reference allele. And so, the reference allele file should contain:
1,2,0,1
This section define how NGSphy is executed. If the user has access to a computational cluster, the ART commands can be converted into jobs for SGE or SLURM schedulers (see Section 7.6.2.). If desired, ART calls can be made by NGSphy transparently to the user (sequentially or in parallel - multi-threading).
[execution]
environment=bash
runART=on
running_times=off
threads=4
-
environment:- purpose: specify in which environment the ART runs are going to be executed (more details below)
- type: enumerate (possible environments).
- values:
- bash: generates a bash file with all the commands used to call ART.
- sge: generates the necessary files to run a job array in a cluster environment running Sun Grid Engine. Includes: seed file, job script and a possible script to launch ART jobs.
- slurm: generates the necessary files to run a job array in a cluster environment running Simple Linux Utility for Resource Management. Includes: seed file, job script and a possible script to launch ART jobs.
-
threads:- purpose: number of threads to execute NGSphy.
- type: number (integer).
- value: 1 (default).
-
runART:- purpose: indicate whether the user actually wants to generate NGS reads This will only run on local, under bash environment
- type: boolean
- values:
- 1, true, on: run ART.
- 0, false, off: don't run ART, bash scripts will be generated (default)
-
runnintg_times:- purpose: obtain the running times file for the NGS mode processes (read counts or ART).
- type: boolean
- values:
- 0, false, off: don’t generate file (default).
- 1, true, on: generate file
IMPORTANT: the generation of this file increases the execution time of the program.
NOTES
- If the execution block is missing, a bash script will be generated and ART instances will not be run.
- If the option environment is missing, a bash script will be generated (default behavior) and ART instances will not be run, unless
runARToption is set. - If the option runART is missing, ART instances will not be run.
- If the value chosen for the option run is wrong and bash is the value of environment, then ART instances will not be run.
- If the value chosen for the option environment is wrong, behavior will be as if there was no execution section, bash script will be generated and ART instances will not be run.
NGSphy can generate job templates for execution in computational clusters running Sun Grid Engine (Gentzsch 2001, Oracle Corp.) or Simple Linux Utility for Resource Management (Yoo et al. 2003, https://slurm.schedmd.com/).
In this case, NGSphy generates two files: project.XXX.sh (job script) and project.seedfile.txt(seed-file for job arrays).
Where:
- XXX will be sge or slurm according to the selected execution environment.
- project: if using any of the single gene tree input modes, will be NGSphy. For the gene-tree distribution input mode, it will be the name of the SimPhy output folder.
To execute this one would type a different command depending on job scheduler (SGE or SLURM)
- SGE:
qsub -t 1-100 project.sge.sh
- SLURM:
sbatch --array 1-100 project.slurm.sh
Here there are some arbitrary examples of the files generated:
- SEED FILE
art_illumina art_illumina -ss GA2 -amp -p -sam -na -i /home/user/NGSphy_output/individuals/1/01/testwsimphy_1_1_data_0.fasta -l 50 -f 10 -o /home/user/NGSphy_output/reads/1/01/testwsimphy_1_1_data_0_R
art_illumina art_illumina -ss GA2 -amp -p -sam -na -i /home/user/NGSphy_output/individuals/1/01/testwsimphy_1_1_data_1.fasta -l 50 -f 10 -o /home/user/NGSphy_output/reads/1/01/testwsimphy_1_1_data_1_R
art_illumina art_illumina -ss GA2 -amp -p -sam -na -i /home/user/NGSphy_output/individuals/1/01/testwsimphy_1_1_data_2.fasta -l 50 -f 10 -o /home/user/NGSphy_output/reads/1/01/testwsimphy_1_1_data_2_R
...
- SGE job script:
#!/bin/bash
# SGE submission options
#$ -l num_proc=1 # number of processors to use
#$ -l h_rt=00:10:00 # Set 10 mins - Average amount of time for up to 1000bp
#$ -t 1-{0} # Number of jobs/files that will be treated
#$ -N art.sims # A name for the job
command=$(awk 'NR==$SGE_TASK_ID{{print $1}}' $SEEDFILE)
$command
- SLURM job script
#!/bin/sh
#SBATCH -n 1
#SBATCH --cpus-per-task 1
#SBATCH -t 00:10:00
#SBATCH --mem 4G
#SBATCH --array=1-1000
command=$(awk 'NR==$SLURM_ARRAY_TASK_ID{{print $1}}' $SEEDFILE)
$command
IMPORTANT: Take into account that the job script files generated by NGSphy are general templates, and that in most cases they will be have to be modified according the the particular cluster environments. It is strongly encouraged to consult the cluster administrator for proper execution.
Generated to keep track of the timings for each ART call or each NGS read counts process. File name follows the format:
project.info
where, project will be NGSphy, if using any of the single gene tree input modes. Whereas, for the gene-tree distribution input mode, it will be the name of the SimPhy output folder.
Content of the file is formatted as follows:
repID,locID,indID,inputFile,cpuTime,seed,outputFilePrefix
-
repID: replicate identifier. -
locID: locus identifier. -
indID: individual identifier. -
inputFile: path of the input file, corresponding to the individual FASTA file. -
cpuTime: processing time -
seed: if the NGS mode needs a seed for the generation of random numbers, it will be here. -
outputFilePrefix: prefix of the file generated.
The output of NGSphy will depend on the NGS mode selected (ngs-read-counts or ngs-reads-art). In both cases, the user will get a detailed log file and a folder structure as:

FIGURE 12: Folder structure of the NGSphy output.
Folder structure include:
- alignments: for single gene tree modes, stores the alignments and files generated for the INDELible run.
- coverage: stores tables describing the coverage for each locus and individual, one per replicate.
-
individuals: stores the FASTA files with the individual sequences. Structured along the hierarchy
replicate > locus > individuals. - ind_labels: stores the correspondence between sequences and individuals.
-
reads: for Illumina reads, stores the ALN/BAM and/or FASTQ files generated by ART. For read counts, stores all the VCF files. Structured as hierarchy
replicate > locus > ALN/BAM/FASTQ/VCF files - ref_alleles: stores the sequences of the references alleles used for the simulation of read counts.
- scripts: stores all the bash scripts generated.
Stores the simulated sequences in FASTA format, both unaligned (*.FASTA) and aligned (*_TRUE.fasta) together with INDELible/INDELible-NGSphy control file, ancestral sequence, and gene trees.
alignments/
|__ngsphy.tree # if inputmode = 3
|__NGSphy.indelible.times # if running_times=1
|__1
|__control.txt
|__ancestral.fasta # if inputmode in [2,3]
|__ngsphydata_1.fasta
|__ngsphydata_1_TRUE.fasta
|__LOG.txt # default indelible file
|__tree.txt # default indelible file
NOTE: During the simulation process, the way it is implemented, the anchor sequence in the alignment produced by INDELible might include indels.
This folder will contain two types of files, comma-separated value (CSV) files with the coverage distribution of each individual per replicate, and plain text files with information of the coverage heterogeneity (multipliers). Format of the filenames are:
| Coverage | Multipliers |
|---|---|
| project.repID.coverage.csv | project.repID.multipliers.txt |
Where:
- project: if using any of the single gene tree input modes, it will be NGSphy. For the gene-tree distribution input mode, it will be the name of the SimPhy output folder.
- repID: number of the replicate.
Folder structure will look like this:
coverage/
|__SimPhyOutput.1.coverage.csv
|__SimPhyOutput.1.multipliers.txt
|__SimPhyOutput.2.coverage.csv
|__SimPhyOutput.2.multipliers.txt
...
The coverage files, one per replicate, store a matrix of shape (number of individuals X number of loci) where each cell corresponds to the depth of coverage of the loci for the specific individual.
indID ,L.01 ,L.02 ,L.03 ,L.04
0 ,4.317, 401.540, 467.337, 0.000
1 ,7.646, 711.199, 827.737, 0.0
2 ,6.401, 595.411, 692.976, 0.0
3 ,10.602, 986.243, 1147.850, 0.0
4 ,15.316, 1424.710, 1658.165, 0.00
The multipliers files, also one per replicate, stores all the multipliers involved in the coverage calculations. These files have 9 sections, starting with a “#” symbol and the name of the section. The sections, ordered, are:
- Experiment coverage: shows the expected coverage per replicate.
- Locus alpha shapes: shows the alpha shapes sampled that will be used for the Gamma distribution that will introduced the coverage variation among loci.
- Individual alpha shapes: shows the alpha shapes sampled that will be used for the Gamma distribution that will introduced the coverage variation among individuals.
- Locus multipliers: the sampled multipliers for the loci.
- Individual multipliers: the sampled multipliers for the individuals.
- On target loci: ID of the loci that are considered as on target.
- Off target loci: ID of the loci that are considered as off target.
- Not captured loci: ID of the loci that are considered as not captured.
- Taxon decay: this will match the given taxon specific parameters from the settings file.
An example of this file:
# Experiment coverage:
[100.0]
# Locus alpha shapes:
0.927251162511
# Individual alpha shapes:
1.53083011312
# Locus Multipliers:
1 0.834697014116
2 1.18198759526
3 0.65934212521
4 0.678000738762
5 0.473412350281
# Individual Multipliers:
1 0.259996456042
2 0.816753359147
3 0.165530070861
4 1.57630793049
5 1.3641198608
# On target loci
1,3,4
# Off target loci
2
# Not captured loci
5
# Taxon decay:
1,0.5
2,0.25
These will store the correspondence between the original sequences and the generated individuals. Each table is a CSV file named as follows:
project.repID.individuals.csv
Where:
-
project: if using any of the single gene tree input modes, it will be NGSphy. For the gene-tree distribution input mode, it will be the name of the SimPhy output folder. -
repID: number of the replicate.
Folder structure will look like this:
ind_labels/
|__NGSphy.1.individuals.csv
This folder will contain tables with the correspondence between the individual identifier and the corresponding sequence identifier. CSV file format:
repID, indID, spID,locID,geneID
1, 0, 0, 0, 0
1, 1, 1, 0, 1
1, 2, 1, 0, 2
Where:
-
repID: identifier of the replicate to which the gene trees and sequences belong. -
indID: identifier of the haploid individual. -
spID: identifier of the species -
locID: identifier of the locus -
geneID: identifier of the gene tree tip.
These tables will contain the correspondence between each individual and its two sequences. CSV file format:
repID,indID,spID,locID,mateID1,mateID2
1, 1, 1, 0, 3, 0
1, 2, 1, 0, 4, 1
1, 3, 1, 0, 2, 5
1, 4, 3, 0, 4, 0
Where:
-
repID: identifier of the replicate. -
indID: identifier of the generated diploid individual. -
spID: identifier of the species. -
locID: identifier of the locus. -
mateID(1&2): identifier of the gene tree tip used for the1(2)sequence of the individual.
This folder will store the diploid individual sequence files (i.e., 2 sequences for each locus), hierarchically organized within replicates and loci. For example:
individuals/
|__1/
|__1/
|__prefix_1_1_ind1.fasta
|__prefix_1_1_ind2.fasta
|__prefix_1_1_ind3.fasta
|__2/
|__prefix_1_2_ind1.fasta
|__prefix_1_2_ind2.fasta
|__prefix_1_2_ind3.fasta
|__2/
|__1/
|__prefix_2_1_ind1.fasta
|__prefix_2_1_ind2.fasta
|__prefix_2_1_ind3.fasta
|__2/
|__prefix_2_2_ind1.fasta
|__prefix_2_2_ind2.fasta
|__prefix_2_2_ind3.fasta
This folder contains the FASTA files with the reference allele sequences used in the VCF file with the read counts. Folder is structured per replicate. There is a reference allele file per locus. Each file contains a single sequence. The format of each file name:
project_REF_repID_locID.fasta
Where:
-
project: if using any of the single gene tree input modes, it will beNGSphy. For the gene-tree distribution input mode, it will be the name of the SimPhy output folder. -
repID: replicate identifier. -
locID: locus identifier.
Folder structure will look like this:
ref_alleles/
|__1/
|__NGSphy_REF_1_1.fasta
|__NGSphy_REF_1_2.fasta
...
|__2/
|__NGSphy_REF_2_1.fasta
|__NGSphy_REF_2_2.fasta
...
This folder will store all the scripts for ART execution, according to the options in the execution block. If we decide to run NGSphy for any cluster environment, we will have the job script and the seed file. If we choose bash as environment and we do not want to execute the ART commands within NGSphy, we would have a single bash script.
SGE
> reads/
|__project.sge.sh
|__project.sh
SLURM
> reads/
|__project.slurm.sh
|__project.sh
BASH
> reads/
|__project.sh
Where, project will be NGSphy, if using any of the single gene tree input modes. Whereas, for the gene-tree distribution input mode, it will be the name of the SimPhy output folder.
Data will be structured per replicate.
This folder will store the output of ART. It follows the same folder structure of the individuals folder, but instead of having FASTA files, it will contain the FASTQ files [and alignment and mapping files (ALN and SAM) if requested ] generated by ART.
reads/
|__1/
|__1/
|__prefix_1_1_ind1_R1.fq
|__prefix_1_1_ind1_R2.fq
|__prefix_1_1_ind2_R1.fq
|__prefix_1_1_ind2_R2.fq
|__2/
|__prefix_1_2_ind1_R1.fq
|__prefix_1_2_ind1_R2.fq
|__prefix_1_2_ind2_R1.fq
|__prefix_1_2_ind2_R2.fq
|__2/
|__1/
|__prefix_2_1_ind1_R1.fq
|__prefix_2_1_ind1_R2.fq
|__prefix_2_1_ind2_R1.fq
|__prefix_2_1_ind2_R2.fq
|__2/
|__prefix_2_2_ind1_R1.fq
|__prefix_2_2_ind1_R2.fq
|__prefix_2_2_ind2_R1.fq
|__prefix_2_2_ind2_R2.fq
NOTE: Independently of the environment chosen and the value of the “runART” option, NGSphy will generate the hierarchical folder structure.
This folder will store the output obtained from the read count simulation. This folder is structured
in 2 sub-folders (with and without sequencing errors), each structured per replicate,
and containing as many VCF files as loci.
Sub-folders will be:
-
no_error: VCF files with the simulated read counts without sequencing error. -
with_error: VCF files with the simulated read counts with the introduced sequencing error.
reads
|__no_error/
|__1/
|__prefix_1_1_TRUE.VCF
|__prefix_1_2_TRUE.VCF
|__prefix_1_3_TRUE.VCF
|__2/
|__prefix_2_1_TRUE.VCF
|__prefix_2_2_TRUE.VCF
|__prefix_2_3_TRUE.VCF
|__with_error/
|__1/
|__prefix_1_1.VCF
|__prefix_1_2.VCF
|__prefix_1_3.VCF
|__2/
|__prefix_2_1.VCF
|__prefix_2_2.VCF
|__prefix_2_3.VCF
In the output folder there will be a file which contains a summary of the parameters used for the simulation. The name of the file will have the format:
NGSPHY.YYYYMMDD-HH:mm:SS.summary.log
-
YYYY: year -
MM: month -
DD: day -
HH: hours -
mm: minutes -
ss: seconds
Here, an example of the output:
Settings:
[GENERAL]
path : ./
output_folder_name : NGSphy_case1_100x_RC
ploidy : 2 (Diploid individuals)
seed : 50426717
numreplicates : 1
numlociperreplicate : 1
filtered_replicates : 1
numindividualsperreplicate : 8
[DATA]
inputmode : 4 (Gene-tree distribution - SimPhy output)
simphy_folder_path : SimPhy_usecase
simphy_data_prefix : data
[COVERAGE]
experiment : f:100
[NGS-READ-COUNTS]
read_counts_error : 0
reference_alleles_file : files/my_reference_allele_file.case1.txt
[EXECUTION]
environment : bash
threads : 2
runart : off
running_times : 0
Stores information related to the time used in each ART run or read-count thread per locus. This file will contain input/output files for each process and its corresponding individual, locus (gene-tree) and replicate (REPID). See more on Section 7.6.3.
Example of the file:
1,1,0,output/individuals/1/01/test_wrapper_1_01_data_0.fasta, 0.013984, 1479977980,output/reads/1/01/test_wrapper_1_01_data_0_R
1,1,1,output/individuals/1/01/test_wrapper_1_01_data_1.fasta, 0.014757, 1479977980,output/reads/1/01/test_wrapper_1_01_data_1_R
1,1,2,output/individuals/1/01/test_wrapper_1_01_data_2.fasta, 0.013589, 1479977980,output/reads/1/01/test_wrapper_1_01_data_2_R
1,1,3,output/individuals/1/01/test_wrapper_1_01_data_3.fasta, 0.013404, 1479977980,output/reads/1/01/test_wrapper_1_01_data_3_R
1,1,4,output/individuals/1/01/test_wrapper_1_01_data_4.fasta, 0.013775, 1479977980,output/reads/1/01/test_wrapper_1_01_data_4_R
For each NGSphy run is optional to get a debug log file. If the -l/--log option in the command line is set to DEBUG, the file will be generated in the current working directory and under the name:
NGSPHY.YYYYMMDD-HH:mm:SS.log
-
YYYY: year -
MM: month -
DD: day -
HH: hours -
mm: minutes -
ss: seconds
This file stores information of the program execution, at a very detailed level. A debug log file will look like this:
13/08/2017 11:21:19 AM - ERROR (__main__|handlingCmdArguments:82): Something happened while parsing the arguments.
Please verify. Exiting.
Advances in sequencing technologies have now made very common that datasets for phylogenomic inference consist of large numbers of loci from multiple species and individuals. The use of next-generation sequencing (NGS) for phylogenomics implies a complex computational pipeline where multiple technical and methodological decisions are necessary that might influence the final tree obtained, from coverage to assembly, mapping, variant calling and/or phasing. In order to assess the influence of these variables, here we introduce NGSphy, an open-source tool for the genome-wide simulation of Illumina reads obtained from thousands of gene families evolving under a common species tree, with multiple haploid and/or diploid individuals per species, where sequencing coverage (depth) heterogeneity can be modelized across individuals and loci, including off target loci and phylogenetic decay. Moreover, parameter values for the different replicates can be sampled from user-defined statistical distributions.

FIGURE 13: A possible analysis pipeline for multilocus, multispecies datasets with multiple individuals with the final goal of exploring the sensitivity of species tree inferences to NGS parameterization variation.
With NGSphy you can generate:
- haploid individuals from gene-tree distributions
- diploid individuals from gene-tree distributions
- genome sequences of haploid/diploid individuals from a single gene tree
- genome sequences of haploid/diploid individuals from a single gene tree and an user-defined ancestral sequence
- genome sequences of haploid/diploid individuals from a single gene tree, an user-defined anchor sequence and an anchor tip.
- NGS Illumina reads of haploid/diploid individuals
- NGS read counts of haploid/diploid individuals
- For the NGS data generation, variation of coverage due to the following:
- variation across individuals and/or loci
- targeted-sequencing effects
- on/off target loci
- on target loci not captured
- taxon-specific variation
Huang W, Li L, Myers JR, and Marth, GT (2012) ART: a next-generation sequencing read simulator. Bioinformatics 28 (4): 593-594
ART (http://www.niehs.nih.gov/research/resources/software/biostatistics/art/) is a set of simulation tools to generate synthetic next-generation sequencing reads. ART simulates sequencing reads by mimicking real sequencing process with empirical error models or quality profiles summarized from large recalibrated sequencing data. ART can also simulate reads using user own read error model or quality profiles. ART supports simulation of single-end, paired-end/mate-pair reads of three major commercial next-generation sequencing platforms: Illumina’s Solexa, Roche’s 454 and Applied Biosystems’ SOLiD. ART can be used to test or benchmark a variety of method or tools for next-generation sequencing data analysis, including read alignment, de novo assembly, SNP and structural variation discovery. ART outputs reads in the FASTQ format, and alignments in the ALN format. ART can also generate alignments in the SAM alignment or UCSC BED file format.
William Fletcher and Ziheng Yang (2009) INDELible: A flexible simulator of biological sequence evolution. Molecular Biology and Evolution. 26 (8): 1879–88. doi:10.1093/molbev/msp098
INDELible (http://abacus.gene.ucl.ac.uk/software/indelible/) is an application for biological sequence simulation that combines many features. Using a length-dependent model of indel formation it can simulate evolution of multi-partitioned nucleotide, amino-acid, or codon data sets through the processes of insertion, deletion, and substitution in continuous time.
Nucleotide simulations may use the general unrestricted model or the general time reversible model and its derivatives, and amino-acid simulations can be conducted using fifteen different empirical rate matrices. Substitution rate heterogeneity can be modelled via the continuous and discrete gamma distributions, with or without a proportion of invariant sites. INDELible can also simulate under non-homogenous and non-stationary conditions where evolutionary models are permitted to change across a phylogeny. Unique among indel simulation programs, INDELible offers the ability to simulate using codon models that exhibit nonsynonymous/synonymous rate ratio heterogeneity among sites and/or lineages.
Diego Mallo, Leonardo De Oliveira Martins and David Posada (2015). SimPhy : Phylogenomic Simulation of Gene, Locus, and Species Trees. Systematic Biology., November, syv082. doi:10.1093/sysbio/syv082
SimPhy (https://github.com/adamallo/simphy) is a program for the simulation of gene family evolution under incomplete lineage sorting (ILS), gene duplication and loss (GDL), replacing horizontal gene transfer (HGT) and gene conversion (GC). SimPhy simulates species, locus and gene trees with different levels of rate heterogeneity, and uses INDELible to evolve nucleotide/codon/aminoacid sequences along the gene trees. The input for SimPhy are the simulation parameter values, which can be fixed or sampled from user-defined statistical distributions. The output consists of sequence alignments and a relational database that facilitate posterior analyses.

FIGURE 14: NGSphy workflow
NGSphy, verifies all the content of the project, the settings files involved and/or the existence of the corresponding third-party applications in order to run. If the input data corresponds to the single gene tree an user-defined ancestral sequence, first the tree is rooted to the selected gene-tree tip. The next step (for any single gene tree input mode) is to evolve the tree under the specific evolution mode to obtain the expected genome sequences. Then (any input mode), the generation of individuals, whether haploid or diploid:
For haploid individuals, resulting genome sequences are separated into single FASTA files and identified. In addition, a file is generated with the correspondence between the individual generated and the description of the sequence it belongs to. For diploid individuals, there is a process of verification that the project content includes species-trees with an even number of individuals per taxa. Sequences are then "paired", individuals being generated by randomly sampling without replacement two sequences within the same gene family and species. Output will include a table for each replicate with the identifiers for the sequences paired and the individuals generated.
Afterwards, the coverage variation matrices will be computed according to the parameters introduced and finally the sequencing data generated, consist on either Illumina reads or read counts (VCF files).
For the Illumina reads, program calls out ART, the NGS simulator, with the parameters established in the settings file and generates reads from the previously generated individuals. Resulting files depend on the settings introduced, and they are files related to the execution of the ART processes (scripts and text files), and the output of such processes (ALN, BAM and/or FASTQ files). For read counts, two scenarios are simultaneously computed, with and without errors.
The read count approach is based on the assumption (Ritz et al., 2011) that the sequencing process is uniform in generating short reads from the target genome, and that the number of reads mapped to a region is expected to be proportional to the number of times the region appears in a DNA sample (Ji and Chen, 2015). Read counts are produced under a user-defined error rate. First, the variable sites (regarding the reference sequences) are identified. Then, coverage for each position is sampled from a Negative Binomial distribution whose mean and overdispersion parameter are the sampled coverage for the specific locus and individual. For diploid individuals, coverage is further splitted among chromosomes with equal probability. Genotype likelihoods for every site are computed as in GATK (McKenna et al 2010) (see also Korneliussen et al. 2014). The output is a set of VCF files, one per locus.
Most common issues, doubts and questions should be solved by reading this manual. If that is not the case or you find any bug, you can post an issue to this repository for reproducibility purposes, with the following files attached:
- the settings file
-
<simphy_project_name>.commandfile or theindelible_control.txtfile.
This software has been developed for Linux/Mac environments and specifically tested under:
Linux Kernel:
4.8.0-58-generic #63~16.04.1-Ubuntu SMP Mon Jun 26 18:08:51 UTC 2017 x86_64 x86_64 x86_64 GNU/Linux
Distribution:
Ubuntu 16.04.2 LTS
Hardware:
Dual core Intel Core i5-3427U (-HT-MCP-) cache: 3072 KB
8GB RAM
- Bragg JG, Potter S, Bi K and Moritz, C. (2016). Exon capture phylogenomics: efficacy across scales of divergence. Molecular ecology resources, 16(5), 1059-1068.
- Fletcher W and Yang Z. (2009) INDELible: A flexible simulator of biological sequence evolution. Molecular Biology and Evolution. 26 (8): 1879–88.
- Gentzsch W. (2001). Sun grid engine: Towards creating a compute power grid. In Cluster Computing and the Grid, 2001. Proceedings. First IEEE/ACM International Symposium on (pp. 35-36). IEEE.
- Huang W, Li L, Myers JR and Marth, GT. (2012) ART: a next-generation sequencing read simulator. Bioinformatics 28 (4): 593-594
- Ji T and Chen J. (2015) Modeling the next generation sequencing read count data for DNA copy number variant study. Stat. Appl. Genet. Mol. Biol. 2015; 14(4): 361–374.
- Korneliussen TS, Albrechtsen A and Nielsen R. (2014) ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinformatics 15:356.
- Mallo D, De Oliveira Martins L and Posada D. (2016). SimPhy : Phylogenomic Simulation of Gene, Locus, and Species Trees. Systematic Biology 65(2): 334-344.
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M and DePristo MA. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research, 20(9), 1297-1303.
- Ritz A, Paris PL, Ittmann MM, Collins C and Raphael BJ. (2011). Detection of recurrent rearrangement breakpoints from copy number data. BMC Bioinformatics, 12(1), 114.
- Yoo AB, Jette MA and Grondona M. (2003). Slurm: Simple linux utility for resource management. In Workshop on Job Scheduling Strategies for Parallel Processing (pp. 44-60). Springer, Berlin, Heidelberg.